Главная Обратная связь
Дисциплины:
Архитектура (936)
Биология (6393)
География (744)
История (25)
Компьютеры (1497)
Кулинария (2184)
Культура (3938)
Литература (5778)
Математика (5918)
Медицина (9278)
Механика (2776)
Образование (13883)
Политика (26404)
Правоведение (321)
Психология (56518)
Религия (1833)
Социология (23400)
Спорт (2350)
Строительство (17942)
Технология (5741)
Транспорт (14634)
Физика (1043)
Философия (440)
Финансы (17336)
Химия (4931)
Экология (6055)
Экономика (9200)
Электроника (7621)

Физические и химические свойства оксикислот
|
|
Оксикислоты – жидкости или большей частью кристаллические
вещества. В воде они растворимы лучше, чем соответствующие карбоновые
кислоты, не содержащие гидроксила. Низшие оксикислоты смешиваются с
водой в любых соотношениях.
Химическое поведение оксикислот, как и других
гетерофункциональных соединений, то есть соединений, содержащих в
молекуле разные функциональные группы, определяется природой этих
групп. Оксикислоты вступают в большинство химических реакций,
характерных для кислот (-COOH) и спиртов (-OH). Строение
углеводородного радикала также сказывается на их химическом поведении:
ароматические кислоты (например, салициловая кислота) вступают во
многие реакции, характерные для соответствующих производных бензола. В
одних реакциях каждая из функциональных групп может участвовать
независимо друг от друга, в других ход реакции и характер образующихся
продуктов зависит от взаимного влияния этих групп, в некоторых случаях
окси- и карбоксильная группы могут взаимодействовать одновременно или
между собой.
1. Кислотныесвойства.
По сравнению с карбоновыми кислотами с тем же числом углеродных атомов
оксикислоты (в особенности α-кислоты) обнаруживают более сильно
выраженные кислотные свойства. Так, константа диссоциации гликолиевой
кислоты в 8,5 раз больше, чем уксусной.
2. Какспирты.
Вступление в реакцию спиртового или кислотного гидроксила
определяется выбором реагента и условиями реакций. Так, при действии НСl
замещается гидроксил спиртовой, а при действии PCl5 и кислотный и
спиртовой. Во всех превращениях оксикислот приходится считаться с
взаимным влиянием гидроксильной (-ОН) и карбоксильной (-СООН) групп.
3. α-Оксикислотылегковосстанавливаются в карбоновыекислоты.
4. Термическоеповедениеоксикислот.
Отщепление Н2О при температуре происходит легко, причем в
зависимости от положения гидроксильной группы образуются различные
вещества.
28АЛЬДЕГИДОКИСЛОТЫ И КЕТОКИСЛОТЫ,органические соединения, в молекулах к-рых, наряду с присущей кислотам карбоксильной группой
 содержится группа
содержится группа  (альдегидокислоты) либо группа
(альдегидокислоты) либо группа 
(кетокислоты). Примером альдегидокислоты может служить глиоксиловая к-та (I), а кетокислоты - пировиноградная к-та (II).


А. и к. встречаются в природных продуктах, нек-рые из них играют важную роль в обмене веществ, являются промежуточными продуктами при спиртовом брожении Сахаров.
АЛЬДЕГИДЫ,класс органич. соединений, содержащих карбонильную группу
 связанную с органич. радикалом (R) и с атомом водорода,
связанную с органич. радикалом (R) и с атомом водорода,
Свойства А. во многом сходны  со свойствами кетонов, также содержащих карбонильную группу, но связанную с двумя радикалами, R2CO. Назв. "А." обычно производят от назв. соответствующих кислот. Так, муравьиной кислоте НСООН соответствует муравьиный альдегид, или формальдегид НСНО; уксусной кислоте - уксусный альдегид, или ацеталь-дегидСНзСНО. Так, при окислении первичных спиртов или при осторожном восстановлении хлорангидридов кислот образуются А.:
со свойствами кетонов, также содержащих карбонильную группу, но связанную с двумя радикалами, R2CO. Назв. "А." обычно производят от назв. соответствующих кислот. Так, муравьиной кислоте НСООН соответствует муравьиный альдегид, или формальдегид НСНО; уксусной кислоте - уксусный альдегид, или ацеталь-дегидСНзСНО. Так, при окислении первичных спиртов или при осторожном восстановлении хлорангидридов кислот образуются А.:

Промежуточному положению А. отвечает и их способность к реакциям окисления-восстановления; напр., в присутствии спиртового раствора едкой щёлочи А. превращаются в смесь спирта и кислоты

А. могут быть получены также пиролизом смешанных кальциевых солей муравьиной и к.-л. другой карбоновой к-ты:

Осторожным окислением ароматич. соединений, содержащих метильную группу,получают ароматич. А.

Технич. значение имеет аналогичный способ получения простейшего ненасыщенного А.- акролеина - из пропилена:

Метод синтеза ацетальдегида, имеющий пром. значение, состоит в гидратации ацетилена в присутствии солей ртути (см. Кучерова реакция):

А. склонны к полимеризации; формальдегид, напр., легко превращается в пара-формальдегид, ацетальдегид - в цик-лич. тример, т. н. паральдегид. При конденсации 2 молей А. образуются альдоли:

(см. Альдольная конденсация), к-рые с отщеплением воды могут образовать ненасыщенные альдегиды:

Еще химсв-ва :
Гидрирование (присоед водорода) по двойной связи карбонильной группы легко протек при пропускании паров оксосоед над Ni или Pd катализатором. При этом кетоны образ вторич спирты а альжегиды первичные :
Присоединение реактивов Гриньяра по двойной связи карбонильной группы
Присоединение HCN к карбонильной группе альдегидов и кетонов
Присоединение Н2О характерно только для наиболее активных альдегидов
Присоединение спиртов
Присоединение бисульфата
Замещение карбонильного кислорода на галоген
Р-я галогенирования
Альдольная и кротоновая конденсации оксосоедин
Простейшая альдегидокислота – глиоксалевая кислота HC(=O)–COOH – может быть получена гидролизом дихлоруксусной кислоты или электролитическим восстановлением щавелевой кислоты. Она встречается в природе в незрелых плодах. Простейшая -кетокислота – пировиноградная кислота CH3COCOOH – является важным промежуточным веществом в окислительном и ферментативном расщеплении сахаров (в гликолизе) живыми организмами. Ее можно получить, используя общие реакции, ведущие к образованию -кетокислот:

-Кетокислоты легко окисляются:

Они также очень чувствительны к действию концентрированной серной кислоты, вызывающей реакцию

-Кетокислоты. Свободные -кетокислоты при нагревании легко выделяют углекислый газ, образуя кетоны:

Однако их эфиры вполне устойчивы. Их можно получить из сложных эфиров конденсацией Клайзена:

Их простейший представитель – ацетоуксусный эфир CH3COCH2COOC2H5 – имеет большое значение для органического синтеза. Его натриевая соль CH3–CO–CHNa–COOC2H5, получаемая действием этилата натрия, легко реагирует с алкилгалогенидами, давая алкилацетоуксусные эфиры, CH3COCHRCOOC2H5. Их в свою очередь можно проалкилировать до диалкилпроизводных CH3COCRRCOOC2H5. Расщепление таких эфиров до замещенных уксусных кислот можно осуществить действием сильных щелочей:

Расщепление до замещенных ацетонов достигается обработкой разбавленными кислотами или щелочами:

29. Липиды- сложные эфиры трехатомного спирта глицерина в высших карбоновых кислот. По строению их подразделяют на сложные и простые. Твердые жиры- обычно глицериды предельных к-т: миристиновой( С14), пальмитиновой (С16) и т д. Жидкие жиры- сложные эфиры непред к-т: чаще всего олеиновой.
Химическиесв-васложных липидов
Как к-ты, благодаря наличию карбокс групп в сос-ве молекул, они реагируют со щелочами, спиртами, аммиаком и образуют при этом соли, эфиры, амиды и т д
Как спирты, благодаря наличию спиртового гидроксила, оксикислотыреагир с металлич натром, образуя алкоголяты.
Гидролиз в зав-ти от условий бывает:
-водный( без катализ-ра, при высоких t , P)
-кислотный ( в присут кислоты в кач-ве катализатора)
- ферментативный
-щелочной
Р-я присоеднинения водорода
Присоединение галогенов
Ф-ии жиров в организме. Энергетическая, Структурная, Защитная.
Липиды входят в состав или служат источником многих важных веществ.
Липиды — важный пластический материал
Способы получения
30.Еще в XIX веке было отмечено, что некоторые органические вещества в жидком состоянии и в растворе способны вращать плоскость поляризации плоскополяризованного света (оптическая активность), причем одни отклоняют ее вправо, а другие – влево. Эта способность впервые была обнаружена Пастером в 1848 г. у винных кислот. С тех пор это явление называется оптической изомерией. Сейчас известно, что оптическая активность связана с наличием в молекуле sp3-гибридизированного атома углерода, связанного с четырьмя различными заместителями. Такой атом углерода называется асимметрическим или хиральным.
Так, например, в молекуле молочной кислоты содержится асимметрический атом углерода (хиральный атом углерода обозначают С*), связанный с четырьмя различными группами.

Наличие асимметрического атома углерода приводит к образованию двух зеркальных изомеров. Они отличаются друг от друга, как предмет от своего изображения в зеркале (как левая и правая руки).

Если тетраэдрические модели молочной кислоты спроектировать на плоскость, то получаются проекционные формулы, названные формуламиФишера по имени ученого, предложившего их:

При использовании формул Фишера нужно помнить, что верхняя и нижняя групп ы (для молочной кислоты это -СООН и -СН3) лежат за плоскостью чертежа, а боковые (для молочной кислоты это -Н и -ОН) – перед плоскостью чертежа. Асимметрический атом углерода находится на пересечении прямых, связывающих эти группы. Такие модели нельзя совместить при вращении в плоскости чертежа (они относятся друг к другу, как предмет к своему зеркальному изображению или как левая и правая руки). Подобные пространственные изомеры называются зеркальными изомерами, энантиомерами или оптическими антиподами. Общее число стереоизомеров N вычисляется по формуле N=2n, где n – количество С*.


31. Моносахариды – это сахароподобные, сладкие на вкус и не подверженные гидролизу до более простых соединений индивидуалного в-ва.Альдозы – многоатомные спирты с альдегидной функциональной группой у первого углеродного атома. По кол-ву атомов углерода альдозы подразделяют: альдотриозы, альдотетрозы, альдопентозы, альдогексозы. Пространственное строение углеводов иллюстрирует существование глицеринового альдегида в виде двух антиподов D (+) L(-) , отличающ расположением спиртового гидроксила у асиметричного атома углерода :
Если строение монозы выводить из глицеринового альдегида, то отнесение любого углевода к D или Lгенетич ряду определ конфигурацией его предпоследнего ассиметрического атома углерода : так, если гидроксил находится справа, то это D –ряд, а если гидроксил слева – L ряд.
Циклические формы альдопетоз и гексопентоз
Циклические формы
При этом открытые и закрытые формы находятся в динамическом равновесии
Пример на маннозе
Кетозы- общее название моносахаридов, содерж помимо гидроксильных еще и кетонную С=О группировку в сос-ве молекулы и способных реагировать как кетонополиспирты. Это излмерныеальдозам соединения, которые по числу атомов углерода подразд: кетотетрозы, кетопентозы, кетогексозы. Кетозысодерж на один ассиметрич атом углерода меньше альдоз. Подобно альдозам принадлежность к D или Lгенетич ряду определ положением спиртового гидроксила у предпоследнего, считая с функцион С=О группы, ассиметрич атома углерода. Эритрулоза :
Кетозы, подобно альдозам, в растворах сущ как в открытой , так и в циклич форме, которые наход в динамич равновесии по отношению друг к другу. На примере D –фруктозы
32. . Моносахариды – это сахароподобные, сладкие на вкус и не подверженные гидролизу до более простых соединений индивидуалного в-ва. Альдозы – многоатомные спирты с альдегидной функциональной группой у первого углеродного атома. По кол-ву атомов углерода альдозы подразделяют: альдотриозы, альдотетрозы, альдопентозы, альдогексозы. Кетозы- общее название моносахаридов, содерж помимо гидроксильных еще и кетонную С=О группировку в сос-ве молекулы и способных реагировать как кетонополиспирты. Это излмерныеальдозам соединения, которые по числу атомов углерода подразд: кетотетрозы, кетопентозы, кетогексозы.
Химсв-ва:
1. Реакционноспособностьполуацетального гидроксила.
2. Р-я алкилирования
3. Эпимеризация моноз
4. Р-я ацилирования
5. Восстановление моноз
33.

Окисление в кислой среде
Окисление аммиачным р-ом оксида серебра
Вопрос 33. Монасахариды-сахароподобные,сладкие на вкус и не подверженные гидролизу до более простых соединений индивидуальные в-ва.
Вопрос 34.Дисахариды- углеводы, кот сод-ат 2 остатка моносахаридов,связанных между собой простой эфирной гликозидной связью.
Строение:

Мальтоза(2 остатка альфа-гликопиранозаа)тип связи 1,4-альфа. Мальтоза-природный дисахарид,встречающийся как продукт неполного гидролиза крахмала.
 сахароза-смешанный дисахарид,остаток глюкозы и фруктозы,тв.кристаллич в-во,сладкий на вкус,синтезируют только растения,раствор в воде.(1,2-альфа,бетта)
сахароза-смешанный дисахарид,остаток глюкозы и фруктозы,тв.кристаллич в-во,сладкий на вкус,синтезируют только растения,раствор в воде.(1,2-альфа,бетта)
 целлобиоза-в природе встречается как продукт неполного гидролиза целлюлозы.
целлобиоза-в природе встречается как продукт неполного гидролиза целлюлозы.

Хим св-ва:1.гидролиз до моносахаридов
2.восстанавливающие св-ва(только дисахаиды имеющие свободн полуацетальный гидроксил:мальтоза,целлобиоза,лактоза,но не сахароза)
3.образование гликозидов(восстанавливающие дисахарды)
Все гликозил-гликозы имеют свобод.полуацетальный гидроксил и относятся к восстанавливающим дисахаридам(восстанавливают реактив Фелинга и серебро из аммиачного р-ра его оксида). К наиболее часто встречающимся восстанавли-им дисах-ам относятся:альфа-1,4-мальтоза,бетта-1,4-целлобиоза и лактозы,альфа-1,6-меллибиоза. Их восстанав-ие св-ва обусловлены способностью образовывать в растворах открытые формы.
К невосстанавливающим дисахаридам(гликозил-гликозидам) следует отнести трегалозу и сахарозу(см выше ее строение). Невосстанавливающие сахара не имеют ОН-группы ни при одном аномерном центре, восстанавливающие – имеют свободную ОН-группу при аномерном центре.
Вопрос 35.Образование из 2-х моносахаридов дисахарид В-мальтозу.(глюкоза+глюкоза=мальтоза)
 +
+ 

Природный дисахарид состоящий из двух остатков глюкозы, является восстанавливающим сахаром, так как имеет незамещённую полуацетальную гидроксильную группу,мальтоза восстанавливает фелингову жидкость,способна к мутаротации.
 образование восстанав-его дисах-да
образование восстанав-его дисах-да
Вопрос 36.полисахариды-высокомолекулярные полимерные углеводы,построенные из большого числа остатков моноз,соединенных между собой гликозид-гликозной связью. Если полисах-ды сост из остатков одного моносах-да,их относят к гомополисах-ам(крахмал,целлюлоза),гетерополисах-ды-сополимеры,в состав кот входят различные монозы.
Полисах-ды-бесцветные,аморфные в-ва,большинство кот вообще не растворимо в воде;они выполняют роль конструкционного материала клеток растительных организмов(целлюлоза). Водорастворимые же полиозы(например,крахмал,гликоген) явл.запасными в-ми в растительных и животных организмах.
Крахмал-природная смесь 2-х полисахаридов(20% амилоза и 80% амилопектин). Строение амилозы-линейный полисах-д,сод-щий остатки альфа-глюкопиранозы,тип связи 1,4-альфа.,растворим в воде.
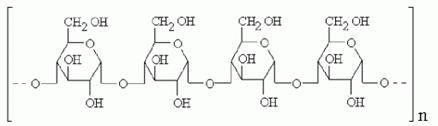
Строение амилопектина-сильно разветвленный полисахарид,не растворим в воде,также образованный из остатков альфа-глюкопиранозы,тип св. 1,4-альфа, в точке ветвления тип связи 1,6-альфа.

Крахмал- главное резервное пит-ое в-во растений,в виде зерен содержится во многих органах растений,особенно запасающих.представляет собой белый аморфный порошок,не раствор в спирте,хол.воде,эфире.
Целлюлоза-природный полисах-д из остатков В-глюкопиранозы. В чистом виде изестна в виде хлопка и фильтрованной бумаги. Много(до 50%)клетчатки сод-ся в древисине. Не растворим в воде,только набухает,ее способны переваривать только некот виды организмов(жвачные животные до40% и термитные 85%) . ее синтезируют только растения,волокнистый материал,оч прочный и с трудом подвергается гидролизу.

Хим св-ва: 1. Гидролиз(300градусов,серная к-та,Р) целлюлозы протекает ступенчато до глюкозы через промежуточные низкомолекул-ые целлодекстрины: целлюлоза---(везде над стрелками Н2О и Н+)---целлодекстрины---целлобтоза---глюкоза.
2.р-ии по ОН-гр для получения искусственного волокна(ацетатный шелк) (см лекцию)
Вопрос 37. Ами́ны— органические соединения, являющиеся производными аммиака, в молекуле которого один, два или три атома водорода замещены на углеводородные радикалы.Классиф-ия:первичные R-NH2,вторичныеR-R-NH,третичные R-R-R-N: Номенклатура: К названию органических остатков, связанных с азотом, добавляют слово «амин» при этом группы упоминают в алфавитном порядке: CH3NHC3H7 — метилпропиламин, CH3N(C6H5)2 — метилдифениламин,СН3-NH2-метиламин,С6Н5-NH2-фениламин(анилин). Основные виды изомерии по строению R
Методы получения:1.Взаимодействие аммиака со спиртами NH3+CH3OH(Al2O3,t над стрелкой,под стр. –Н2О)--СН3NH2(метиламин) СН3ОН+СН3NН2СН3(СН3)-NН-диметиламин и т.д до (СН3)3N 2.Р-ия ГофманаI(последовательное взаимод-ие галогенуглеводородов с NH3): а) СН3С1(хлорметан)+NH3 CH3NH3+ * C1-(солянокислый метиламин) б)CH3NH3+ * C1- +СН3С1(н.стр+NH3) NH4Cl+ (CH3)2NH2+ *C1-(солянокисл диметиламин) в)(CH3)2NH2+ *C1-+ СН3С1 NH4Cl+ (CH3)3NH+ *C1-(солянокислый триметиламин) г)т.д. получится тетраметиламмонийхлористый 3.Р-ия ГофманаII (окисление амидов кислот)

амид уксусной к-ты
4.последовательное взаим-ие ROH c NH3 NH3+CH3OH(Al2O3,t над стрелкой,под стр. –Н2О)--СН3NH2(метиламин) СН3ОН+СН3NН2СН3(СН3)-NН-диметиламини т.д до (СН3)3N
На основность аминов влияют различные факторы: электронные эффекты углеводородных радикалов,способность образующихся ионов к стабилизации за счет сольватации в среде растворителя. В результате +I-эффекта алкильных групп основность алифатических аминов в газовой фазе (без растворителя) растет в ряду: первичные < вторичные < третичные.

Вопрос 38. Ами́ны— органические соединения, являющиеся производными аммиака, в молекуле которого один, два или три атома водорода замещены на углеводородные радикалы.Классиф-ия:первичные R-NH2,вторичныеR-R-NH,третичные R-R-R-N:
Хим св-ва: 1. Ацилирование(введение остатка карб-ой к-ты R-C-=O)

2. р-ия алкилирования 
3. Солеобразования (взаимод-ие с кис-ми) 1.СН3NH2(метиламин)+ НС1CH3-NH3+ * Cl-(cолянокисл метиламин) 2.СН3-NH2+HOSO3HCH3-NH3+*OSO3H-(серно-кислая соль метиламина)
4.взамодействие аминов с HNO3: а)первичные амины:СН3-NH2+HO-N=OCH3-OH+ H2O+N2 б)вторичные амины превращаются в нитрозаамины: (СН3)2NH(диметиламин)+HO-N=O (н.стр.НС1,Н2О) Н2О+ (СН3)2-N-N=O (N-N-диметилнитрозоамин)желтый р-р. 3) третичные амины-р-ии нет. 4)ароматического амина 
Вопрос 53
Пиридин.
Общая характеристика
Гетероциклические соединения — органические соединения, содержащие в своих молекулах циклы, в образовании которых принимают участие неуглеродные атомы (гетероатомы). Гетероциклические соединения классифицируют по числу атомов в цикле и по типу гетероатома. В данной главе мы рассмотрим только некоторые азотсодержащие гетероциклы, производные которых имеют важное биохимическое значение.
Шестичленные гетероциклы
Пиридин C5H5N - простейший шестичленный ароматический гетероцикл с одним атомом азота. Его можно рассматривать как аналог бензола, в котором одна группа СН заменена на атом азота:

Строение. По электронному строению пиридин напоминает бензол. Все атомы углерода и атом азота находятся в состоянии sp2-гибридизации. Шесть электронов (по одному от каждого атома), находящихся на негибридных орбиталях, образуют p-электронную ароматическую систему. Из трех гибридных орбиталей атома азота две вступают в образование σ-связей C-N, а третья содержит неподеленную пару электронов .
Физические свойства. Пиридин — бесцветная жидкость, немного легче воды, с характерным неприятным запахом; с водой смешивается в любых отношениях. 
Получение. Пиридин выделяют из каменноугольной смолы, в которой его содержание 0,08%. В лабораторных условиях пиридин можно синтезировать из синильной кислоты и ацетилена:
2HC≡CH + HC≡N  C5H5N.
C5H5N.
Химические свойства
Химические свойства пиридина определяются наличием ароматической системы и атома азота с неподеленной электронной парой.
1. Основные свойства. Пиридин — более слабое основание, чем алифатические амины (Кb = 1,7.10-9). Его водный раствор окрашивает лакмус в синий цвет:

При взаимодействии пиридина с сильными кислотами образуются соли пиридиния:

2. Ароматические свойства. Подобно бензолу, пиридин вступает в реакции электрофильного замещения, однако его активность в этих реакциях ниже, чем бензола, из-за большой электроотрицательности атома азота. Пиридин нитруется при 300 °С с низким выходом:

Атом азота в реакциях электрофильного замещения ведет себя как заместитель 2-го рода, поэтому электрофильное замещение происходит в мета-положение.
В отличие от бензола, пиридин способен вступать в реакции нуклеофильного замещения, поскольку атом азота оттягивает на себя электронную плотность из ароматической системы, и орто-пара-положения по отношению к атому азота обеднены электронами. Так, пиридин может реагировать с амидом натрия, образуя смесь орто- и пара-аминопиридинов (реакция Чичибабина):

3. При гидрировании пиридина образуется пиперидин, который представляет собой циклический вторичный амин и является гораздо более сильным основанием, чем пиридин:

4. Гомологи пиридина по свойствам похожи на гомологи бензола. Так, при окислении боковых цепей образуются соответствующие карбоновые кислоты:

Никотиновая кислота и ее амид — важные лекарственные препараты
Пиримидин
Пиримидин C 4H 4N 2 - шестичленный гетероцикл с двумя атомами азота. Его можно рассматривать как аналог бензола, в котором две группы СН заменены на атомы азота:

Благодаря наличию в кольце двух электроотрицательных атомов азота пиримидин еще менее активен в реакциях электрофильного замещения, чем пиридин. Его основные свойства также выражены слабее, чем у пиридина.
Основное значение пиримидина состоит в том, что он является родоначальником класса пиримидиновых оснований.
Производные пиримидина широко распространены в живой природе, где участвуют во многих важных биологических процессах. В частности, такие производные как цитозин, тимин, урацил входят в состав нуклеотидов, являющихся структурными единицами нуклеиновых кислот, пиримидиновое ядро входит в состав некоторых витаминов группы B, в частности B1, коферментов и антибиотиков.
Ÿ 
Ÿ Структурная формулацитозина
Ÿ
Ÿ
Ÿ 
Ÿ Структурная формулатимина
Ÿ
Ÿ
Ÿ 
Ÿ Структурная формулаурацила
Ÿ
Пиримидиновая структура — как ароматическая, так и гидрированная, входит в состав многих биологически активных веществ и лекарственных препаратов — например, барбитуратов — производных 1,3,5-тригидроксипиридина, обладающих снотворным, противосудорожным и наркотическим действием.
Вопрос 54.
Индол (1-H-indole) — органическое соединение ряда азотсодержащих гетероциклов. Является родоначальником широкого класса природных соединений.

Состоит из конденсированных бензольного (6 атомов углерода) и пятичленного пиррольного ядра, содержащего в цикле один атом азота.
Химические свойства
Индол - слабое основание (рКа —2,4). При протонировании образует катион 3H-индолия (формулла I), который при взаимодействии с нейтральной молекулой индола дает димер (II).

Индол, подобно пирролу, мало устойчив к действию кислот, поэтому реакции с электрофилами проводят, избегая сильнокислых сред.
Преимущественное направление электрофильной атаки для индола – положение 3 гетероцикла, что находится в соответствии с эффективной резонансной стабилизацией образующегося при этом s-комплекса. Отметим, что в случае атаки по положению 2 резонансная стабилизация s-комплекса с участием гетероатома возможна только через делокализацию положительного заряда по бензольному кольцу, что менее выгодно вследствие нарушения ароматичности всей системы.

Примерами реакций индола, протекающих как электрофильное замещение, являются реакция Вильсмаейра, приводящая к 3-формилиндолу, и аминометилирование – реакция Maннuxa – взаимодействие с формальдегидом и солями вторичных аминов.
При взаимодействии формальдегида с вторичным амином в кислой среде образуется соль N,N-диметилметилидениминия, которая и выступает в роли электрофила. Надо отметить, что реакция Манниха характерна не только для индола и других p-избыточных гетероциклов, но и для многих СН-активных соединений, например кетонов.
В случае индола реакция с формальдегидом и хлоргидратом диметиламина приводит к грамину – алкалоиду, встречающемуся в некоторых растениях. Более важным является то обстоятельство, что грамин является исходным соединением в промышленном синтезе триптофана – незаменимой аминокислоты. С этой целью грамин вводят во взаимодействие с натриевой солью эфира ацетамидомалоновой кислоты, образующийся малоновый эфир гидролизуют и декарбоксилируют. Подробнее об этом можно прочесть в пособии, посвященном аминокислотам.

Еще одно полезное вещество, которое получают по реакции электрофильного замещения в индоле, это индолилуксусная кислота или гетероауксин – стимулятор роста растений. Гетероауксин образуется при взаимодействии индола с Na-солью хлоруксусной кислоты в щелочной среде и последующем подкислении реакционной смеси для выделения свободной кислоты. Это реакция алкилирования, то есть электрофильного замещения в ароматическом ядре, однако условия ее протекания принципиально отличаются от условий, характерных для реакции Фриделя-Крафтса. Возможность ее протекания связана с тем, что индол, подобно пирролу, является достаточно сильной NH-кислотой, способной депротонироваться с образованием аниона. Последний, будучи анионом, гораздо легче реагирует с электрофилом, чем исходный индол, и электрофильная атака направлена в положение 3.

Приведенный выше пример показывает, что в реакции электрофильного замещения зачастую вводят не сам индол, а его анион, который обычно генерируют действием сильного основания, например магнийорганического соединения. В отличие от аниона пиррола в этом случае реакции протекают в несколько более жестких условиях и всегда по положению 3. Еще один пример такого рода превращения это взаимодействие индола с амилнитритом в присутствии метилата натрия, которое приводит к оксиму.

Следует отметить, что во всех случаях электрофильное замещение в индоле происходит по положению 3. Если положение 3 занято электронодонорным заместителем, то реакция протекает по положению 2, и только если в положении 3 находится электроноакцепторный заместитель, реакция может идти по бензольной части молекулы. Это означает, что если нужно синтезировать индол, замещенный в бензольном кольце, необходимо чтобы заместитель уже присутствовал на стадии построения гетероцикла. Например, 4-оксизамещенные индолы могут быть получены взаимодействием циклогександиона-1,3 с a-бромкетонами и последующей реакцией с первичными аминами илиаммиаком.

Как слабая кислота (рKа 17), индол с Na в жидком NH3 образует N-натрийиндол, с КОН при 130°С - N-калийиндол.
Обладает ароматическими свойствами. Электрофильное замещение идет главным образом в положение 3.
Нитрование обычно осуществляется бензоилнитратом, сульфирование - пиридинсульфотриоксидом, бромирование - диоксандибромидом, хлорирование - SO2Cl2, алкилирование - активными алкилгалогенидами.
Ацетилирование в уксусной кислоте также идет в положение 3, в присутствии CH3COONa - в положение 1; в уксусном ангидриде образуется 1,3-диацетилиндол. Индол легко присоединяется по двойной связи a,b-непредельных кетонов и нитрилов, например:

Взаимодействует с альдегидами приводит к образованию дииндолильных производных, например:

Аминометилирование (реакция Манниха) в мягких условиях протекает в положение 1, в жестких - в положение 3. Замещение в бензольное кольцо (преимущественно в положения 4 и 6) идет лишь в кислых средах при блокированном положении 3.
В присутствии Н2О2, надкислот или на свету индол окисляется в индоксил, который затем превращается в тример или индиго. Более жесткое окисление под действием О3, МnО2 приводит к разрыву пиррольного кольца с образованием 2-формамидобензальдегида. При гидрировании индола водородом в мягких условиях восстанавливается пиррольное кольцо, в более жестких - и бензольное, например:

Нахождение в природе
Содержится в каменноугольной смоле, в некоторых эфирных маслах (например, в масле жасмина и цитрусовых).
Индол встречается в некоторых эфирных маслах (жасминовом, апельсиновом), входит в состав гликозида индикана, природной аминокислоты триптофана и других соединений.
Индольная система типична для растений чилибухи, спорыньи, калабарских бобов, раувольфии и других растений, содержащих алкалоиды.
Вопрос 55
Пурин — простейший представитель имидазо[4,5-d]пиримидинов. Бесцветные кристаллы, хорошо растворимые в воде, горячем этаноле и бензоле, плохо растворимые в диэтиловом эфире, ацетоне и хлороформе.  Производные пурина играют важную роль в химии природных соединений (пуриновые основания ДНК и РНК; кофермент NAD;алкалоиды, кофеин, теофиллин и теобромин; токсины, сакситоксин и родственные соединения; мочевая кислота) и, благодаря этому, в фармацевтике.
Производные пурина играют важную роль в химии природных соединений (пуриновые основания ДНК и РНК; кофермент NAD;алкалоиды, кофеин, теофиллин и теобромин; токсины, сакситоксин и родственные соединения; мочевая кислота) и, благодаря этому, в фармацевтике.
|
Просмотров 4338 |
|
|
