Главная Обратная связь
Дисциплины:
Архитектура (936)
Биология (6393)
География (744)
История (25)
Компьютеры (1497)
Кулинария (2184)
Культура (3938)
Литература (5778)
Математика (5918)
Медицина (9278)
Механика (2776)
Образование (13883)
Политика (26404)
Правоведение (321)
Психология (56518)
Религия (1833)
Социология (23400)
Спорт (2350)
Строительство (17942)
Технология (5741)
Транспорт (14634)
Физика (1043)
Философия (440)
Финансы (17336)
Химия (4931)
Экология (6055)
Экономика (9200)
Электроника (7621)

Застигання гарячого розчину желатину веде до утворення холодцю
|
|
Закон Гесса
В основі термохімічних розрахунків лежить закон Гесса: Тепловий ефект (ΔН) хімічної реакції (при постійних Р і Т) залежить від природи і фізичного стану вихідних речовин та продуктів реакції і не залежить від шляху її протікання.
Наслідки із закону Гесса:
1. Теплові ефекти прямої і зворотної реакцій рівні за величиною і протилежні за знаком.
2. Тепловий ефект хімічної реакції (ΔН) дорівнює різниці між сумою ентальпій утворення продуктів реакції та сумою ентальпій утворення вихідних речовин, взятих з урахуванням коефіцієнтів у рівнянні реакції (тобто помножені на них).
Закон Гесса може бути записаний у вигляді наступного математичного виразу:
 .
.
За допомогою закону Гесса можна розрахувати ентальпії утворення речовин і теплові ефекти реакцій, які неможливо виміряти експериментально.
1.3. Закон Кірхгофа
Закон Кірхгофа встановлює залежність теплового ефекту хімічної реакції від температури: температурний коефіцієнт теплового ефекту хімічної реакції дорівнює зміні теплоємності системи в ході реакції. Закон Кірхгофа лежить в основі розрахунку теплових ефектів при різних температурах.
4. Закон Гесса дозволяє обчислити теплові ефекти процесів, для яких відсутні експериментальні дані. Це стосується не тільки хімічних реакцій, але й процесів розчинення, випарювання, сублімації, кристалізації та ін. При термохімічних розрахунках особливо значимі два види теплових ефектів: ентальпія утворення та ентальпія горіння сполук.
Ентальпія утворення сполук є тепловим ефектом реакції утворення одного моля даної сполуки із простих речовин за стандартних умов. Наприклад, стандартна ентальпія утворення DfН карбонату кальцію – це тепловий ефект реакції
Са(т) + С(графіт) + 1,5О2(г) = СаСО3(т), DfН = - 1206 кДж.
При цьому ентальпія утворення простих речовин (Н2, О2, Са, С та ін.) дорівнює нулю, а ентальпії утворення більшості відомих речовин можна відшукати у довідниках.
За значною кількістю стандартних ентальпій утворення можна обчислити теплові ефекти багатьох хімічних реакцій. При цьому використовують правило, яке випливає із закону Гесса:
тепловий ефект хімічної реакції дорівнює різниці суми ентальпій утворювання кінцевих речовин та суми ентальпій утворювання вихідних речовин із урахуванням коефіцієнтів, що подані перед позначенням речовин у рівнянні реакції.
Нехай хімічна реакція проходить відповідно до рівняння
аА + bВ = сС + dD DН - ?
Тут а, b, с, d –коефіцієнти перед речовинами А, В, С, D.Тоді DН = (сDfНС + dDfНD) – ( aDfНA + bDfНB).
Для наочності розглянемо конкретний приклад. Реакцію горіння етану С2Н6 описується рівнянням
С2Н6(г) + 3,5О2(г) = 2СО2(г)+3Н2О(ж),
DН298 =-1559,87 кДж/моль.
Обчислити ентальпію утворення етану, якщо відомі ентальпії утворення вуглекислого газу та води: DfН298(СО2)= -393,51 кДж/моль, DfН298 (Н2О) = - 285,84 кДж/моль.
Відповідно до закону Гесса маємо:
DН = 2DfН(СО2) + 3DfН(Н2О) - DfН(С2Н6).
Звідси DfН298(С2Н6) = 2DfН298(СО2) + 3DfН298(Н2О) - DН298 ==2(-393,51) + 3(-285,84) – (-1559,87) = -84,67 кДж/моль.
Ентальпією згоряння сполуки називають тепловий ефект реакції окислення даної сполуки киснем за стандартних умов з утворенням вищих оксидів елементів, що входять до складу цієї сполуки. Наприклад, стандартна ентальпія згоряння DСН етилового спирту – це тепловий ефект реакції
С2Н5ОН(ж) + 3О2 = 2СО2(г) + 3Н2О(ж).
Продуктами сгоряння є СО2, Н2О(г) або Н2О(ж), SO3 та інші. Якщо серед продуктів реакції крім оксидів, наявні інші речовини (наприклад, N2, HCl), то це спеціально обумовлюється. Ентальпії згоряння вищих оксидів та інших продуктів сгоряння, а також кисню брати за нуль. За допомогою ентальпій згоряння можна також розрахувати теплові ефекти хімічних реакцій, використовуючи таке правило:
тепловий ефект хімічної реакції дорівнює різниці суми ентальпій згоряння вихідних речовин та сумі ентальпій згоряння продуктів реакції з урахуванням коефіцієнтів, що подані перед позначенням речовин у рівнянні реакції.
5.Дру́гий закон термодина́міки — один із основних законів фізики, закон про неспадання ентропії візольованій системі. Він накладає обмеження на кількість корисної роботи, яку може здійснити тепловий двигун. На засадничому рівні другий закон термодинаміки визначає напрямок протікання процесів у фізичній системі - від порядку до безпорядку. Існує багато різних формулювань другого закону термодинаміки, загалом еквівалентних між собою. ля системи із сталою температурою існує певна функція стану S — ентропія, яка визначається таким чином, що
1. Адіабатичний перехід із рівноважного стану A в рівноважний стан B можливий лише тоді, коли
 .
.
2. Приріст ентропії у квазістаціонарному процесі дорівнює
 ,
,
де T — температура.
Формулюваняя:
Неможливо перетворити теплоту в роботу, не виконуючи ніякої іншої дії крім охолодження системи
Неможливо створити вічний двигун 2-го роду
Самочинний перехід теплоти від менш нагрітого до більш нагрітого неможливий
Вільна енергія Гіббса (або просто енергія Гіббса, або потенціал Гіббса, або термодинамічний потенціал у вузькому сенсі) - це величина, що показує зміну енергії в ході хімічної реакції і дає таким чином відповідь на принципову можливість протікання хімічної реакції; це термодинамічний потенціал такого вигляду: 
Второй закон термодинамики объясняет направление
протекания процессов и вводит понятие энтропии ΔS=Q/T.
Критерием самопроизвольного протекания процесса в
изолированной системе является ΔS>0, равновесия – ΔS=0.
2. Энтропия является функцией состояния и ее смысл
расшифровывается в статистической термодинамики как мера
беспорядка системы. Чем больше беспорядок, тем больше
энтропия.
3. Критериями самопроизвольного протекания процессов при p,
T=const является ΔG<0; V, T=const - ΔF<0. Критериями
равновесия – ΔG=0 при p, T=const и ΔF=0 при V, T=cons
Расчитывая ΔG и ΔF мы можем оценить только возможность
(термодинамическую вероятность) протекания процесса.
При реальном предсказании возможности протекания процесса надо
учитывать еще и скорость его протекания
6.Хімічна рівновага - стан хімічної системи, в якому оборотно протікає одна або кілька хімічних реакцій, причому швидкості в кожній парі пряма-зворотна реакція рівні між собою. Для системи, що перебуває в хімічному рівновазі, концентрації реагентів, температура і інші параметри системи не змінюються з часом.[1]
А 2 + В 2 ⇄ 2AB
1. Зміщення хімічної рівноваги
Положення хімічної рівноваги залежить від наступних параметрів реакції: температури, тиску і концентрації. Вплив, який чинять ці фактори на хімічну реакцію, підпорядковуються закономірності, яка була висловлена в загальному вигляді в 1885 французьким ученим Ле-Шательє.
Фактори що впливають на хімічну рівновагу:
1) температура
При збільшенні температури хімічна рівновага зміщується в бік ендотермічний (поглинання) реакції, а при зниженні в сторону екзотермічної (виділення) реакції.
CaCO3 = CaO + CO2-Q t ↑ →, t ↓ ←
N2 +3 H2 ↔ 2NH3 + Q t ↑ ←, t ↓ →
2) тиск
При збільшенні тиску хімічна рівновага зміщується в бік меншого обсягу речовин, а при зниженні в бік більшого об'єму. Цей принцип діє тільки на гази, тобто якщо в реакції беруть участь тверді речовини, то вони в розрахунок не беруться.
CaCO3 = CaO + CO2 P ↑ ←, P ↓ →
1моль = 1моль +1 моль
3) концентрація вихідних речовин та продуктів реакції
При збільшенні концентрації одного з вихідних речовин хімічна рівновага зміщується в бік продуктів реакції, а при збільшенні концентрації продуктів реакції-убік вихідних речовин.
S2 +2 O2 = 2SO2 [S], [O] ↑ →, [SO2] ↑ ←
Каталізатори не впливають на зміщення хімічної рівноваги! До екзергонічних реакцій належать катаболічні реакції - реакції розщеплення або окиснення “паливних” молекул (енерговмісних нутрієнтів), котрі надходять в організм у складі харчових продуктів.
Доендергонічних реакцій належать анаболічні реакції - реакції синтезу складних біоорганічних сполук – клітинних макромолекул.
В біології міцно закріпилось уявлення, що дихання в організмі (окиснення) за своєю суттю є процесом “горіння", яке відбувається дуже повільно. Отже, поживні речовини, що потрапили в організм з їжею, є “паливом", яке згорає в організмі шляхом приєднання кисню повітря. Разом з тим, було звернуто увагу на той факт, що повільне “горіння" органічних речовин в організмі істотно відрізняється від такого, що проходить поза організмом: по-перше, воно відбувається при низькій температурі, по-друге, – при відсутності полум'я і, по-третє, – за наявності води, вміст якої в тканинах досить високий.
Біологічне окиснення – це сума всіх окисно-відновних процесів, включаючи анаеробні, що відбуваються в клітинах організму (цитоплазмі, мітохондріях, ендоплазматичному ретикулумі). Основними субстратами біологічного окиснення є вуглеводи та ліпіди, саме їх катаболізм дає найбільшу кількість хімічної енергії, що акумулюється у високоенергетичних зв’язках макроергів.
Функції біологічного окиснення:
1. Забезпечення клітин енергією;
2. Забезпечення клітин пластичним матеріалом для відтворення структур організму;
3. Знешкодження токсичних речовин.
7.Залежність швидкості реакції від концентрації реагуючих речовин. Залежність швидкості реакції від концентрації реагуючих речовин виражається основним законом хімічної кінетики – законом діючих мас (ЗДМ):
швидкість гомогенної хімічної реакції за постійної температури прямо пропорційна добутку концентрацій реагуючих речовин, узятих в степенях їхніх стехіометричних коефіцієнтів в рівнянні реакції.
Для реакції aA + bB = cC + dD математичний вираз закону запишеться так:
Ѵгом = kСАа×СВb, (5.1)
де Ѵгом – швидкість реакції; k – константа швидкості хімічної реакції; САа і СВb – концентрації реагуючих речовин, моль/л; а, b – стехіометричні коефіцієнти в рівнянні реакції.
Фізичний зміст константи швидкості (k): k показує, з якою швидкістю відбувається реакція за концентрацій реагуючих речовин 1 моль/л. Константа швидкості залежить від природи реагуючих речовин, температури, присутності каталізатора, але не залежить від концентрації реагуючих речовин і парціальних тисків (для газів).
Для реагуючих речовин в газоподібному стані замість концентрацій в законі діючих мас можна використати їх парціальні тиски:
Ѵгом = k×рАа×рВb. (5.2)
У разі гетерогенних процесів в закон діючих мас входять концентрації тільки тих речовин, які знаходяться в газовій фазі або розчині. Концентрації речовин, що знаходяться в твердій фазі, постійні і включені в константу швидкості. Наприклад:
C(т) + О2(г) = СО2(г) 
Zn(т) + 2HCl(ж) = ZnCl2(ж)+ H2(г) 
константою рівноваги хімічної реакції називається добуток рівноважних концентрацій кінцевих речовин, поділений на добуток рівноважних концентрацій початкових речовин, причому всі концентрації взяті у ступенях, які однакові із стехіометричними коефіцієнтами реакції.
Константа рівноваги не залежить від концентрацій початкових і кінцевих речовин, але залежить від природи речовин і температури.
способи її вираження: через парціальні тиски (Кр), через концентрації (Кс), через мольні частки (Кх)
принципу Ле Шательє:
якщо на систему, що перебуває в стані рівноваги, чиниться який-небудь зовнішній вплив (змінюється концентрація, температура, тиск), то він сприяє перебігу тієї з двох протилежних реакцій, яка послаблює цей вплив.
якщo нa cucmeмy, щo nepeбyвaє y cmaнi piвнoвaгu, noдiяmu ззoвнi, mo в cucmeмi вiдбyвamимymьcя змiнu, щo nocлaблююmь абo знuщyюmь цю дiю.
Cиcтeмa пepeйдe з oднoro cтaнy piвнoвaги в iнший, який вiдпoвiдaтимe нoвим yмoвaм. Цe пoв'язaнo з тим, щo зoвнiшня дiя piзнoю мipoю змiнює швидкicть двox пpoтилeжнo нaпpямлeниx пpoцeciв.
Poзглянeмo вплив piзниx чинникiв нa cтaн xiмiчнoї piвнoвaги.
Bплив кoнцeнтpaцiї нa cтaн piвнoвaги. Згiднo з пpинципoм Лe-Шaтeльє, ввeдeння в cиcтeмy, щo пepeбyвaє в cтaнi piвнoвaги, дoдaткoвoї кiлькocтi бyдь-якoї з peaгyючиx peчoвин викликaє змiщeння piвнoвaги y тoмy нaпpямкy, в якoмy її кoнцeнтpaцiя змeншyєтьcя. Ocь чoмy дoбaвляння в cиcтeмy oднiєї з виxiдниx peчoвин cпpичинює змiщeння piвнoвaги впpaвo, a дoбaвляння пpoдyктiв peaкцiї— влiвo.
Bплив тиcкy нa cтaн piвнoвaги. Для гaзoвиx cиcтeм нa cтaн xiмiчнoї piвнoвaги впливaє тиcк, ocкiльки iз збiльшeнням тиcкy зpocтaє кoнцeнтpaцiя гaзoвиx кoмпoнeнтiв y дaнiй cиcтeмi. Peaкцiї, щo cyпpoвoджyютьcя змeншeнням oб'ємy, лeгшe йдyть зa пiдвищeнoгo тиcкy. Oтжe, згiднo з пpинципoм Ле-Шaтeльє, пiдвищeння тиcкy зyмoвлює змiщeння xiмiчнoї piвнoвaги в нaпpямкy пpoцecy, який cyпpoвoджyєтьcя змeншeнням oб'ємy, a знижeння тиcкy — викликaє змiщeння piвнoвaги y пpoтилeжний бiк. Oтжe, нaпpямoк змiщeння piвнoвarи визнaчaєтьcя знaкoм DV. У paзi oбчиcлeння DV мoжнa знexтyвaти oб'ємoм нeгaзoпoдiбниx peaгeнтiв
Bплив тeмпepaтypи нa cтaи piвнoвaги. Згiднo з пpинципoм Лe-Шaтeльє, пiд чac нaгpiвaння cиcтeми, щo пepeбyвaє в cтaнi piвнoвaги, ocтaння змiщyєтьcя в бiк тогo з двox пpoтилeжнo нaпpямлeниx пpoцeciв, який cyпpoвoджyєтьcя пoглинaнням тeплoти. Пpиpoднo, щo знижeння тeмпepaтypи зyмo
влює npoтилeжний peзyльтaт: piвнoвaгa змiщyєтьcя в бiк тогo пpoцecy, який cyпpoвoджyєтьcя видiлeнням тeплoти. Oтжe, нaгpiвaння cпpияє пepeбiгyeндoтepмiчнoro, a oxoлoджeння — eкзoтepмiчнoгo пpoцecy..
8.Швидкість хімічної реакціївизначається кількістю речовини, що прореагувала за одиницю часу в одиниці об’єму.
Формула середньої швидкості хімічної реакції:

де  — середня швидкість хімічної реакції,
— середня швидкість хімічної реакції,  — зміна концентрації реагенту,
— зміна концентрації реагенту,  — час.
— час.
Чинники, що впливають на швидкість хімічної реакції
1) Природа реагуючих речовин.
2) Агрегатний стан реагуючих речовин.
3) Концентрація реагуючих речовин.
Основний закон хімічної кінетики —закон діючих мас для швидкості хімічних реакцій: швидкість хімічної реакції за сталої температури пропорційна добутку концентрацій реагуючих речовин.
Для реакції  :
:
 ,
,
де k — коефіцієнт;  і
і  — концентрації реагуючих речовин (моль/л).
— концентрації реагуючих речовин (моль/л).
4) Площа поверхні зіткнення реагуючих речовин.
Ця залежність справедлива для гетерогенних систем за участю твердих речовин.
5) Зміна температури.
Залежність швидкості реакції від температури описуєтьсяправилом Вант-Гоффа: при підвищенні температури на кожні 10 °С швидкість більшості реакцій збільшується у 2—4 рази.
 ,
,
де  — температурний коефіцієнт.
— температурний коефіцієнт.
6) Для газів — тиск у системі.
7) Наявність каталізаторів.
Каталізатори — це речовини, що змінюють швидкість хімічної реакції. Каталізатор у процесі реакції не витрачається і до складу кінцевих продуктів не входить.Позитивний каталіз — прискорення реакції.Негативний каталіз,абоінгібування,— уповільнення реакції.
Константа швидкості реакції (питома швидкість реакції) - коефіцієнт пропорційності в кінетичному рівнянні.
Фізичний сенс константи швидкості реакції k випливає з рівняння закону діючих мас : k чисельно дорівнює швидкості реакції при концентрації кожного з реагуючих речовин дорівнює 1 моль / л.
Константа швидкості реакції залежить від температури, від природи реагуючих речовин, але не залежить від їх концентрації.
9.Залежно від механізму всі хімічні реакції класифікують на прості (елементарні) і складні. Простими називаються реакції, які відбуваються в одну стадію за рахунок одночасного зіткнення молекул, записаних в лівій частині рівняння. У простій реакції можуть брати участь одна, дві або, що зустрічається вкрай рідко, три молекули. Тому прості реакції класифікують на мономолекулярні, бімолекулярний і трімолекулярние реакції. Так як з точки зору теорії ймовірності одночасне зіткнення чотирьох і більше молекул малоймовірно, реакції більш високою, ніж три, молекулярної не зустрічаються. Для простих реакцій кінетичні рівняння відносно прості. Наприклад, для реакції H 2 + I 2 = 2 HI кінетичне рівняння має вигляд
 = K ∙ C (I 2) ∙ C (H 2).
= K ∙ C (I 2) ∙ C (H 2).
Складні реакції протікають у кілька стадій, причому всі стадії пов'язані між собою. Тому кінетичні рівняння складних реакцій більш громіздкі, ніж простих реакцій. Наприклад, для складної реакції H 2 + Br 2 = 2 HBr відомо
 =
=  .
.
Складність кінетичного рівняння безпосередньо пов'язана зі складністю механізму реакції.
Основним законом хімічної кінетики є постулат, що випливає з великого числа експериментальних даних і виражає залежність швидкості реакції від концентрації. Цей закон називають законом діючих мас. Він стверджує, що швидкість хімічної реакції в кожен момент часу пропорційна концентрацій реагуючих речовин, зведеним до деяких ступеня.
Послідовні реакції – це реакції із проміжними стадіями:  Початок реакції, коли речовину D ще не можна зафіксувати, називається періодом індукції. Загальна швидкість реакції визначається швидкістю найбільш повільної (лімітуючої) стадії
Початок реакції, коли речовину D ще не можна зафіксувати, називається періодом індукції. Загальна швидкість реакції визначається швидкістю найбільш повільної (лімітуючої) стадії
До паралельних відносяться спряжені реакції: 
Перша реакція – первинна – протікає спонтанно, а друга – вторинна – тільки під час першої. Речовина (  ), яка бере участь в обох процесах, називається актором. Другий учасник (
), яка бере участь в обох процесах, називається актором. Другий учасник (  ) первинної реакції називається індуктором, а другий учасник вторинної реакції (
) первинної реакції називається індуктором, а другий учасник вторинної реакції (  ) – акцептором. Відношення кількостей актора, реагуючих із індуктором та акцептором, називається фактором індукції, величина якого приблизно дорівнює цілому числу. Це свідчить про утворення у ході спряжених реакцій проміжних нестійких речовин. Під час спряжених реакцій концентрація індуктора може: a) зростати (автокаталітичні реакції); b) не змінюватись (каталітичні реакції); c) зменшуватись.
) – акцептором. Відношення кількостей актора, реагуючих із індуктором та акцептором, називається фактором індукції, величина якого приблизно дорівнює цілому числу. Це свідчить про утворення у ході спряжених реакцій проміжних нестійких речовин. Під час спряжених реакцій концентрація індуктора може: a) зростати (автокаталітичні реакції); b) не змінюватись (каталітичні реакції); c) зменшуватись.
Реакції, що протікають під впливом чи УФ- називаються фотохімічними. Енергія активації фотохімічних реакцій обумовлена поглинанням молекулами фотонів світла з утворенням реакційноздатних частинок. Фотосинтез, зір (фоторецепція) – приклади двох важливих біологічних процесів. Це наслідок того, що основним джерелом енергії на Землі є сонячна радіація.
При поглинанні світла молекули переходять в електронно-збуджений стан. Це супроводжується зміною фізичних і хімічних властивостей молекул порівняно із основним станом. Змінюється дипольний момент, геометрія, розподіл електронної густини. Молекула в збудженому стані має іншу реакційну здатність.
Найбільш поширеними фотохімічними реакціями є: фотодисоціація, фотоприєднання, фотовідновлення, фотоокиснення, фотозаміщення, фотоізомерізація.
10.Сума показників степенів у рівнянні швидкості хімічної реакції (так зване кінетичне рівняння) (3) є важливою характеристикою механізму процесу і називається порядком хімічної реакції. Якщо порядок реакції нульовий (швидкість не залежить від концентрації реагуючих речовин), то v = соnst.
Швидкість реакції першого порядку описують кінетичним рівнянням
v = kС.
Прикладом реакцій першого порядку є розкладання оксиду азоту (V):
N2О5 = 2NО2 + 1/2О2.
Для реакцій другого порядку кінетичне рівняння має вигляд
v = kС2; або v = kС1С2 .
Прикладом реакцій другого порядку є взаємодія водню і йоду за рівнянням
Н2 + І2 = 2НІ
і розкладання оксиду азоту (IV):
2NО2 = 2NО + О2 .
Реакції третього порядку описуються кінетичними рівняннями
v = kС3; v = kС12С2; v = kС1С22; v = kС1С2С3 .
Прикладом таких реакцій є:
2NО + О2 = 2NО2;
2NО + Вr2 = 2NОВr.
Порядок реакції, тобто сума показників степенів у кінетичному рівнянні, може набувати і дробових значень.
Для характеристики механізму реакцій застосовують поняття молекулярності реакції. Під молекулярністю реакції розуміють кількість молекул, які беруть участь в елементарному акті взаємодії.
Час напівреакцп (напівперетворення) - це проміжок часу, за який концентрація початкових компонентів зменшується вдвічі
для реакцій першого порядку ї1/2 не залежить від с0 , для реакцій другого порядку він обернено пропорційний с0.
11.Залежність швидкості фізико-хімічного процесу від температури приблизно виражається правилом Вант-Гоффа: зі збільшенням температури на кожні 10 градусів швидкість більшості хімічних реакцій зростає приблизно в 2 -4 рази. Математично ця залежність виражається так:
 (5.3)
(5.3)
 (5.4)
(5.4)
де  и
и 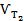 – швидкості реакції за температур Т2 і Т1; ΔТ = Т2 – Т1; γ– температурний коефіцієнт швидкості (значення змінюються від 2 до 4), що показує, в скільки разів збільшиться швидкість реакції з підвищенням температури на 10 градусів. Кількісне значення γ залежить від природи реагуючих речовин і для цієї реакції є величиною сталою.
– швидкості реакції за температур Т2 і Т1; ΔТ = Т2 – Т1; γ– температурний коефіцієнт швидкості (значення змінюються від 2 до 4), що показує, в скільки разів збільшиться швидкість реакції з підвищенням температури на 10 градусів. Кількісне значення γ залежить від природи реагуючих речовин і для цієї реакції є величиною сталою.
Збільшення швидкості хімічної реакції з підвищенням температури пов’язане із зростанням числа активних молекул, що мають надмірну енергію. Для того, щоб брати участь в реакції, молекулам необхідний надлишок енергії в порівнянні з середньою енергією молекул за цієї температури. Цей надлишок енергії називається енергією активації реакції; вона позначається Еа і вимірюється в кДж/моль. Чисельне значення Еа залежить від природи реагуючих речовин і каталізатора. Чим більше значення Еа, тим меншою є швидкість хімічної реакції. Процеси гомогенного каталізу особливо важливі у біохімічних процесах, отже життєдіяльність будь-яких організмів залежить від швидкості переробки продуктів харчування в речовини, які необхідні для розвитку та функціонування організмів. Біологічні каталізатори називають ферментами. Ферменти є речовинами білкового походження. Деякі з них складаються з одного компонента (пепсин, трипсин)
12.
Рівняння Арреніуса встановлює залежність константи швидкості хімічної реакції  від температури
від температури  .
.
Згідно простої моделі зіткнень хімічна реакція між двома вихідними речовинами може відбуватися тільки в результаті зіткнення молекул цих речовин. Але не кожне зіткнення веде до хімічної реакції. Необхідно подолати певний енергетичний бар'єр, щоб молекули почали один з одним реагувати. Тобто молекули повинні володіти певною мінімальною енергією ( енергія активації 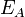 ), Щоб цей бар'єр подолати. З розподілу Больцмана для кінетичної енергії молекул відомо, що число молекул, що володіють енергією
), Щоб цей бар'єр подолати. З розподілу Больцмана для кінетичної енергії молекул відомо, що число молекул, що володіють енергією 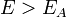 , Пропорційно
, Пропорційно 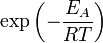 . У результаті швидкість хімічної реакції представляється рівнянням, яке було отримано шведським хіміком Сванте Арреніус з термодинамічних міркувань:
. У результаті швидкість хімічної реакції представляється рівнянням, яке було отримано шведським хіміком Сванте Арреніус з термодинамічних міркувань:

Тут  характеризує частоту зіткнень реагуючих молекул,
характеризує частоту зіткнень реагуючих молекул,  - універсальна газова стала.
- універсальна газова стала.
У рамках теорії активних зіткнень залежить від температури, але ця залежність досить повільна:

Оцінки цього параметра показують, що зміна температури в діапазоні від 200 C до 300 C приводить до зміни частоти зіткнень  на 10%.
на 10%.
У рамках теорії активованого комплексу виходять інші залежності від температури, але у всіх випадках більш слабкі, ніж експонента.
Рівняння Арреніуса стало одним з основних рівнянь хімічної кінетики, а енергія активації - важливою кількісною характеристикою реакційної здатності речовин.
Енергія активації- мінімальна кількість енергії, яке потрібно повідомити системі (в хімії виражається в джоулях на моль), щоб відбулася реакція
У хімічній моделі, відомої як Теорія активних зіткнень (ТАС), є три умови, необхідних для того, щоб відбулася реакція :
Молекули повинні зіткнутися. Це важлива умова, однак його не достатньо, тому що при зіткненні не обов'язково відбудеться реакція.
Молекули повинні володіти необхідною енергією (енергією активації). У процесі хімічної реакції взаємодіючі молекули повинні пройти через проміжне стан, який може володіти більшою енергією. Тобто молекули мають подолати енергетичний бар'єр; якщо цього не відбудеться, реакція не почнеться.
Молекули повинні бути правильно орієнтовані відносно один одного.
При низькій (для певної реакції) температурі більшість молекул мають енергією меншою, ніж енергія активації, і нездатні подолати енергетичний бар'єр. Однак в речовині завжди знайдуться окремі молекули, енергія яких значно вище середньої. Навіть при низьких температурах більшість реакцій продовжують йти. Збільшення температури дозволяє збільшити частку молекул, що володіють достатньою енергією, щоб подолати енергетичний бар'єр. Таким чином підвищується швидкість реакції.
13. Каталіз - виборче прискорення одного з можливих термодинамічно дозволених напрямків хімічної реакції під дією каталізатора, який багаторазово вступає в проміжне хімічна взаємодія з учасниками реакції і відновлює свій хімічний склад після кожного циклу проміжних хімічних взаємодій.
Каталізатор змінює механізм реакції на енергетично більш вигідний, тобто знижує енергію активації. Каталізатор утворює з молекулою одного з реагентів проміжне з'єднання, в якому ослаблені хімічні зв'язки. Це полегшує його реакцію з другим реагентом. Важливо зазначити, що каталізатори прискорюють оборотні реакції, як в прямому, так і в зворотному напрямках..
Каталіз може бути позитивним (коли швидкість реакції збільшується) і негативним (коли швидкість реакції зменшується). Для позначення від'ємного каталізу часто використовують термін інгібування.
Каталіз буває гомогенним і гетерогенним (контактним). У гомогенному каталізі каталізатор складається в тій же фазі, що і реактиви реакції, в той час, як гетерогенні каталізатори відрізняються фазою.
Прикладом гомогенного каталізу є розкладання пероксиду водню в присутності іонів йоду. Реакція протікає у дві стадії:
H 2 О 2 + I → H 2 О + IO
H 2 О 2 + IO → H 2 О + О 2 + I
При гомогенному каталізі дію каталізатора пов'язано з тим, що він вступає у взаємодію з реагують речовинами з утворенням проміжних сполук, це призводить до зниження енергії активації.
При гетерогенному каталізі прискорення процесу зазвичай відбувається на поверхні твердого тіла - каталізатора, тому активність каталізатора залежить від величини і властивостей його поверхні. На практиці каталізатор зазвичай наносять на твердий пористий носій.
Механізм гетерогенного каталізу складніше, ніж у гомогенного. Механізм гетерогенного каталізу включає п'ять стадій, причому всі вони оборотні.
1. Дифузія реагуючих речовин до поверхні твердої речовини
2. Фізична адсорбція на активних центрах поверхні твердого речовини реагують молекул і потім хемосорбция їх
3. Хімічна реакція між реагують молекулами
4. Десорбція продуктів з поверхні каталізатора
5. Дифузія продукту з поверхні каталізатора в загальний потік
Прикладом гетерогенного каталізу є окислення SO 2 в SO 3 на каталізаторі V 2 O 5 при виробництві сірчаної кислоти (контактний метод).
14. Ферменти або ензими - зазвичай білкові молекули або молекули РНК ( рібозіми) або їх комплекси, що прискорюють ( каталізують) хімічні реакції в живих системах. Реагенти в реакції, що каталізується ферментами, називаються субстратами, а отримувані речовини - продуктами. Ферменти специфічні до субстратів (АТФаза каталізує розщеплення тільки АТФ, а киназа фосфорілази фосфорилирует тільки фосфорилазу). Ферментативна активність може регулюватися активаторами і інгібіторами (активатори - підвищують, інгібітори - знижують). Білкові ферменти синтезуються нарибосомах, а РНК - в ядрі.
Ферменти присутні у всіх живих клітинах і сприяють перетворенню одних речовин ( субстратів) в інші (продукти). Ферменти виступають в ролі каталізаторів практично у всіх біохімічних реакціях, що протікають в живих організмах - ними каталізується більше 4000 різних біохімічних реакцій [2]. Ферменти відіграють найважливішу роль у всіх процесах життєдіяльності, направляючи й регулюючи обмін речовин організму.
Подібно до всіх каталізаторів, ферменти прискорюють як пряму, так і зворотну реакцію, знижуючи енергію активації процесу. Хімічна рівновага при цьому не зміщується ні в пряму, ні у зворотний бік. Відмінною особливістю ферментів в порівнянні з небілкових каталізаторами є їх висока специфічність - константа зв'язування деяких субстратів з білком може досягати 10 -10 моль / л і менше. Кожна молекула ферменту здатна виконувати від декількох тисяч до декількох мільйонів "операцій" в секунду. Наприклад, одна молекула ферменту реніну, що міститься в слизовій оболонці шлунка теля, створаживается близько 10 6молекул казеїноген молока за 10 хв при температурі 37 C. При цьому ефективність ферментів значно вище ефективності небілкових каталізаторів - ферменти прискорюють реакцію в мільйони і мільярди разів, небілкові каталізатори - в сотні і тисячі разів
Для кожного ферменту існує своє оптимальне значення рН (рНопт) – це таке рН, при якому швидкість ферментативної реакції оптимальна. При відхиленні від оптимального рН активність ферменту знижується, навіть до мінімуму при значних змінах реакції середовища.
Зміна активності ферменту при різних рН пояснюється:
1) впливом рН на ступінь іонізації функціональних груп в активному центрі ферменту. Саме це індукує конформаційні зміни в активному центрі і відповідно впливає на спорідненість ферменту до субстрату. Окремі групи амінокислотних залишків при різних значеннях рН можуть змінювати заряд і при рНоптзнаходитися в максимально вигідному стані іонізації для каталітичного перетворення субстрату в продукт.
Наприклад:
-NH2 + H+ ↔ -NH4+ або -COOH ↔ -COO- + H+ ;
У ферментативному каталізі так, як і у звичайному хімічному, температура суттєво впливає на перебіг реакції. При підвищенні температури зростає швидкість руху молекул субстрату і, таким чином, імовірність каталітичного перетворення. Але для ферментативного каталізу дуже важливим є вплив температури на конформаційні зміни в молекулі білка-ферменту і, у свою чергу, на структуру активного центру ферменту.
Кожен фермент має оптимальні значення tоС, при яких швидкість реакції максимальна, - це температурний оптимум (tоСопт). На рис. 11 наведений графік залежності активності ферменту від tоС. Для більшості ферментів організму людини від лежить у межах 37-38оС. При зростанні температури вище вказаного температурного оптимуму відбуваються структурні зміни в молекулі ферменту і в його активному центрі, які призводять до зниження активності. Більшість ферментів інактивуються при 40-50оС. Значне підвищення температури викликає навіть денатурацію молекул білка-ферменту з повною втратою каталітичної активності. Для деяких ферментів значне зниження температури може також призвести до повної інактивації.
В умовах надлишку субстрату в реакційному середовищі залежність швидкості реакції від концентрації ферменту має прямо пропорційний характер (рис.12). Тобто в живих клітинах чим більше молекул ферменту, тим швидше відбувається реакція. Цей принцип закладений в основу одного із шляхів регуляції ферментативних процесів, який має назву «зміна кількості ферментів у клітині»
15. Якщо пластинку будь-якого металу, наприклад, цинку занурити у воду, то іони цинку, що утворюють кристалічну решітку металу, під дією полярних молекул води гідратуються, зв'язок їх з решіткою послаблюється, і деяка їх кількість, відриваючись від металу, перейде й воду, а на металі залишиться еквівалентна кількість електронів:
Zn = Zn2+ + 2e-
Між катіонами металу, що перейшли у воду, і негативно зарядженою пластинкою виникає електростатичне притягання, яке зумовлює зворотній процес — перехід іонів металу на пластинку; в системі встановлюється хімічна рівновага.
Іони цинку переходять із пластинки в розчин і осідають з розчину на пластинці з однаковою швидкістю. На межі між металом і розчином утворюється подвійний електричний шар і виникає стрибок потенціалу. Чим міцніше кристалічна решітка металу, тим важче іону металу перейти у розчин. Чим більша величина теплоти гідратації, тим легше іонам перейти у розчин. Отже, при зіткненні металу з водою його йони перебувають під дією двох конкуруючих сил.
Якщо рідина — вода, то для всіх металів в якісному відношенні картина буде однозначною: метал заряджається негативно, шар рідини, що прилягає до нього, позитивно. Інша картина спостерігається у випадку, якщо металеву пластинку занурити в розчин солі цього металу. Якщо метал неактивний, то буде переважати процес осадження іонів з розчину, І пластинка такого металу набуває позитивного заряду.
Отже при зануренні металевої пластинки у розчин власної солі на місці зіткнення металу з розчином виникає стрибок потенціалу, величина і знак якого залежать від хімічної природи металу та від активності його йонів у розчині.
Провідник (метал), занурений у розчин електроліту, називається електродом.
Різниця потенціалів (стрибок потенціалу), що виникає на межі поділу електрод — розчин, називається електродним потенціалом.
Величину електродного потенціалу можна розрахувати за рівнянням Нернста:
Для Т=298 К:
Натепер наука не має у своєму розпорядженні методів, які дозволяють вимірювати абсолютне значення електродних потенціалів, можна виміряти тільки різницю потенціалів. Для цього потрібно якийсь потенціал умовно прийняти рівним нулю. Таким потенціалом є нормальній (стандартний) потенціал водневого електрода. Нормальний водневий електрод являє собою платинову пластинку, покриту платиновою черню, занурену в розчин кислоти, активність іонів Н+ у якому дорівнює 1 моль/л. Через розчин пропускають ретельно очищений водень під тиском 101325 Па (1 ат.). Поверхня платини покривається шаром газоподібного водню. На межі газоподібний водень — іони гідрогену проходить реакція.
У якості стандартного електрода сьогодні користуються не нормальним водневим, а іншим більш простим у виготовленні. Для виміру електродних потенціалів збирають гальванічний елемент — прилад, у якому енергія хімічної реакції безпосередньо перетворюється в електричну енергію. Він складається з двох електродів.
Електрод, потенціал якого визначають, називають Електродом визначення. Інший електрод з відомим значенням електродного потенціалу називають електродом порівняння. ЕРС гальванічного елемента дорівнює максимальній різниці електродних потенціалів. При розрахунках ЕРС від потенціалу позитивного елекроду віднімають потенціал негативного, тому що ЕРС є величиною позитивною.
Значення нормальних (стандартних) потенціалів одержують за умов що електрод визначення занурений у розчин своєї солі з активність іонів 1 моль/л, а вимірювання проходять при Т = 298 К.
16. Нормальний водневий електрод являє собою платинову пластинку, покриту платиновою черню, занурену в розчин кислоти активність іонів Н+ у якому дорівнює 1 моль/л. Через розчин пропускають ретельно очищений водень під тиском 101325 Па (1 ат.). Поверхня платини покривається шаром газоподібного водню. На межі газоподібний водень — іони гідрогену проходить реакція.
У якості стандартного електрода сьогодні користуються не нормальним водневим, а іншим простішим у виготовленні. Для виміру електродних потенціалів збирають гальванічний елемент — прилад, у якому енергія хімічної реакції безпосередньо перетворюється в електричну енергію. Він складається з двох електродів.

17. Відомо, що кристалічна решітка металу складається з позитивно заряджених іонів Мел+ та відносно вільних електронів (електронного газу). При зануренні металічної пластинки у воду під впливом її полярних молекул катіони металу переходять до рідкої фази. На межі «метал — розчин» досить швидко встановлюється рухома окиснювально-відновна рівновага:
Ме + тН20 .—► Ме(Н20)*+ + пе
або спрощено
Ме і—► Мел+ + пе .
Виникає подвійний електричний шар подібний електричному конденсатору великої ємкості. Обкладками конденсатора є з одного боку надлишкові негативні заряди на металі, а з іншого — надлишкові катіони в розчині близько до поверхні металу (рис. 40, а).
Різницю потенціалів, яка виникає між металом та водним середовищем у стані рівноваги, називають рівноважним електродним потенціалом або потенціалом електрода.
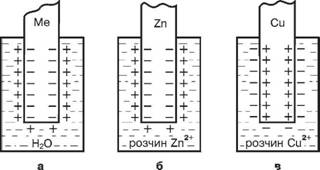
а б в
Рис. 40. Утворення подвійного електричного шару при зануренні: а) металу у воду; б) активного металу у розчин його солі; в) малоактивного металу у розчин його солі
При зануренні металу у розчин його солі виникає подвійний електричний шар, але в цьому випадку можливі два механізми його утворення. Якщо концентрація катіонів металу в розчині не досягає рівноважної або метал досить активний, то рівновага процесу окиснення металу зміщується праворуч, металічна пластинка заряджується негативно (рис. 40, б). Якщо концентрація катіонів
Електродний потенціал — різниця електричних потенціалів між електродом та електролітом, в контакті з яким він знаходиться (найчастіше всього між металом і розчином електроліту).
Якщо пластинку будь-якого металу, наприклад, цинку занурити у воду, то іони цинку, що утворюють кристалічну решітку металу, під дією полярних молекул води гідратуються, зв'язок їх з решіткою послаблюється, і деяка їх кількість, відриваючись від металу, перейде у воду, а на металі залишиться еквівалентна кількість електронів:
Zn = Zn2+ + 2e-
Між катіонами металу, що перейшли у воду, і негативно зарядженою пластинкою виникає електростатичне притягання, яке зумовлює зворотний процес — перехід іонів металу на пластинку; в системі встановлюється хімічна рівновага.
Іони цинку переходять із пластинки в розчин і осідають з розчину на пластинці з однаковою швидкістю. На межі між металом і розчином утворюється подвійний електричний шар і виникає стрибок потенціалу. Чим міцніше кристалічна решітка металу, тим важче іону металу перейти у розчин. Чим більша величина теплоти гідратації, тим легше іонам перейти у розчин. Отже, при зіткненні металу з водою його йони перебувають під дією двох конкуруючих сил.
18.При зануренні металу у воду відбувається відрив іонів від кристалічної гратки під впливом полярних молекул розчинника. В результаті переходу катіонів металу в розчин метал набуває деякого негативного заряду за рахунок електронів, що на ньому залишилися. Прилеглий до металу шар води заряджається позитивно за рахунок катіонів металу. На межі розділення метал - вода утворюється подвійний електричний шар, що і називається електродним потенціалом.
Виникнення потенціалів
При зануренні металу в розчин його солі можливий процес переходу катіонів металу в розчин - тоді метал заряджається негативно.Також можливий перехід катіонів з розчину на метал - тоді метал заряджається позитивно.
Таким чином, кожному металу, зануреному у розчин своєї солі, відповідає той чи інший потенціал. Потенціали, що виникають на металах при зануренні у розчини власних солей, називаються електродними потенціалами. Такі металеві пластинки або дротини називаютьсяелектродами.
Нормальним електродним потенціалом називають потенціал, що виникає на металевій пластинці, зануренній в розчин солі цього ж металу з концентрацією 1 моль еквіваленту в літрі розчину, при порівнянні з потенціалом водневого електрода, потенціал якого прийнято вважати рівним 0.
Стандартным електродом называють напів елемент, що складається з черненої платинової пластинки, насиченої газоподібним воднем при тиску 101,3 кПа (1 атм) і яка знаходиться в розчині іонів водню с активністю, рівною 1 при температурі 298 К
До окисно-відновних електродів належать півелементи, шо складаються з інертного металу (платина, золото), що знаходиться в разчині, що містить окиснену і відновлену форму одного і того ж елемента. Наприклад:Pt/ Fe3+/Fe2+
Інертний метал в цьому електроді не бере участі в електродній реакції, а тільки є переносником електронів. Розчин, що містить одночасно окиснену і відовлену форму речовини, утворює окисно-вілновну систему (редокс-систему).
Потенціал окисно-відновного электрода разраховують за рівнянням Нернста-Петерса:
RT аFe3+
e Fe3+/Fe2+ = e0 + —— lg ———
nF аFe2+
19.
Окисно-відновні реакції мають величезне значення для підтримування життєдіяльності біологічних систем. Процеси фотосинтезу, дихання, травлення – все це ланцюги окисно-відновних реакцій. Ці реакції відіграють чи не найважливішу роль при утворенні власних полімерних компонентів, забезпечуючи організм структурними складовими та енергією.
Проте живий організм є набагато складнішою системою тому і процеси, що в ньому протікають, характеризуються великою складністю, ступінчістю та участю складних ферментативних комплексів.
Живий організм нерозривно пов’язаний з навколишнім середовищем, з якого він одержує необхідні для життя харчові речовини, воду та кисень. З цих речовин в організмі утворюються складні біоорганічні сполуки, які безпосередньо беруть участь у біохімічних перетвореннях.
Життя людини – це складний багатогранний процес, пов’язаний із постійним розщепленням і відновленням в організмі хімічних сполук, що супроводжується витратами енергії. Все це потребує достатнього надходження в організм необхідних неорганічних та органічних речовин, які людина споживає з їжею. Від кількості і якості їжі, режиму харчування залежить рівень функціонального стану, його здоров’я, ріст і розвиток, а також здатність до високопродуктивної розумової і фізичної діяльності.
Енергію, яка витрачається або виділяється в ході окисно-відновних процесів, характеризують
окисно-відновні потенціали. Розрізняють стандартні та формальні, або реальні, окисно-відновні потенціали.
Для реакції, яка відбувається в стандартних умовах (температура 250 С, тиск 101 325 Па (для газоподібних
речовин), концентрації розчинів 1 М), справедливе співвідношення
dG=zFE
де - зміна енергії Гіббса системи;
Е0 – стандартний окисно-відновний потенціал;F – стала Фарадея;.z – число електронів, які приймають участь в процесі
20. Окисно-відновне титрування- група титриметричних методів аналізу, що базуються на використанні окисно-відновних процесів, які є найбільш поширеними і універсальними методами і дозволяють прямо і непрямо визначати неорганічні речовини, а також придатні для визначення багатьох органічних сполук, у т.ч. й фармацевтичних препаратів, переважна більшість яких є потенційними відновниками. Як стандартні розчини застосовують розчини різних окисників або відновників. Залежно від властивостей титранту, який використовують, розрізняють оксидиметрію та редуктометрію. Оксидиметрія — метод визначення відновників шляхом титрування їх стандартними розчинами окисників. Напр. в перманганатометрії як титрант використовують розчин калію перманганату, в броматометрії — калію бромату, в хроматометрії — калію дихромату, в йодометрії — йоду, в йодхлориметрії — йоду монохлориду. Редуктометрія — метод визначення окисників шляхом титрування їх стандартними розчинами відновників. Напр. у гідразинометрії як титрант використовують розчин гідразину гідрогенхлориду, в аскорбінометрії — аскорбінову кислоту, у ферометрії — розчини солей ферум(II)-катіону, в йодометрії — натрію тіосульфату, в нітритометрії — натрію нітриту. Реакції, які застосовують у методі О.-в.т., мають відповідати загальним вимогам до реакцій у титриметричному аналізі, тобто перебіг їх має бути швидким, кількісним, стехіометричним. Але перебіг багатьох реакцій окиснення-відновлення повільний, що пов’язано з їх багатостадійністю. Складний механізм перебігу оксред-реакцій, наявність у системі проміжних продуктів відкривають широкі можливості впливу на їх швидкість зміною умов проведення концентрацій реагентів, температури розчину, введення каталізаторів. Перманганатометрія – окислювачем є перманганат калію, який взаємодіє з окислюючи ми речовинами (відновниками) дуже легко віддає частину свого кисню і марганцем відновлюється. Виділяють 3 атоми кисню, а також кожний атом марганцю приєднує 3 електрони. Тому розрахунок грам – еквівалента перманганата калію буває різноманітним. В першому випадку він дорівнює 31,606 г, а іншому – 58,68 г. Індикатором є сам розчин перманганату калію.Перманганатометрія базується на використанні лікарських речовин, що визначаються, перманганат-іонами. Найчастіше в титриметричному аналіз застосовуються реакції окислення перманганат-іонами в сильно-кислому середовищі. Концентрація кислоти повинна бути не менше 1моль/л. це зумовлено тим, що речовина редокс-потенціалу системи MnO-4/Mn2+ дуже сильно залежить від концентрації кислоти.Для створення кислого середовища застосовують кислоту сульфатну, а не хлороводневі, оскільки хлорид-іони проявляють відновні властивості й можуть бути окислені перманганат-іона до хлору.Нітратна кислота сама є окисником і може викликати побічні реакції, тому її теж не застосовують.Розчин калію перманганату інтенсивно забарвлений у червоно-фіолетовий колір. Навіть 1 крапля 0,01 моль/л забарвлює розчин, що титрується у помітно рожевий колір, тому спеціальних індикаторів у перманганатометрії не застосовують. Нормальний окисно-водний потенціал MnO-4/Mn2+ становить 1,51В, у зв’язку з чим розчин калію перманганату в кислому середовищі можна застосовувати для визначення лікарських речовин, які не взаємодіють з більш слабкими окисниками.Методом перманганатометрії визначають кількісний вміст розчину водню перекису, магнію перекису, натрію нітриту.
21. Йодометрія – це метод кількісного визначення вільного йоду, тих речовин, які кількісно виділяють його під час реакції і тих сполук, які зв’язують його або окислюються йодом у стехіаметричних кількостях.
Йодометричний метод кількісного визначення, має широке практичне застосування і за своєю простотою і точністю він визначається одним із кращих редокс – методів кількісного визначення.
Нормальний окисно-відновний потенціал цієї системи дорівнює 0,545В. Ті речовини, які мають більш низький потенціал, окислюються йодом – іони до йоду, котрий потім може бути від титрований натрію-тіосульфатом.
Пряме йодометричне титрування. Метод прямого йодометричного титрування визначають речовини, які мають сильні відновні властивості (натрію тіосульфат, аскорбінова кислота, лікарські сполуки арсену(ІІІ)та ін.) Визначення проводять у кислому, нейтральному або слабко лужному середовищі. Титр антом є розчин йоду в калію йодиді. Цей розчин має жовто-бурий колір і зайва його крапля забарвлює розчин, що титрується, у блідо-жовтий колір, що може слугувати ознакою кінця титрування (кількісне визначення аналгіну).
Деколи рекомендується додавати декілька літрів органічного розчинника, що не змішується з водою (наприклад хлороформу).
При збовтувані надлишковий йод переходить у хлороформний шар і надає йому фіолетове забарвлення. Однак найближчу чітку кінцеву точку титрування можна визначити за допомогою крохмалю, який з йодом у присутності йодиди-іонів утворює комплексну сполуку інтенсивно сильного кольору.
Методом зворотної йодлметрії визначають сполуки, які повільно окислюються йодом (іоніазид), утворюють з ним комплексні сполуки (кофеїн), дають реакцію ароматичного заміщення (антипірин) або потребують для стехіаметричного необоротного окислення лужного середовища (формальдегід, глюкоза, фурацилін). В останньому випадку окислення відбувається за схемою:
Після завершення реакцій надлишок йоду відтитровують натрію тіосульфатом. Якщо окислення проводили в лужному середовищі до реакційної суміші спочатку додають надлишок кислоти, а тоді йод, що виділяється, відтитровують натрію тіосульфатом.
Крохмаль дають при проведенні зворотного йодометричного визначення у кінці титрування, коли розчин набуде блідо-жовтого кольору – з’явиться інтенсивне синє забарвлення і далі титрують до знебарвлення. Додавати крохмаль до розчинів з великою концентрацією йоду не можна оскільки в цьому випадку відбувається необоротнє зв’язування йоду.
При визначенні речовин, які мають окислюванні властивості (калію перманганат, калію арсенат), до розчину речовини, як правило в кислому середовищі, додають надлишок розчину калію йодиду. У результаті окислювано-відновної реакції виділяється еквівалентна кількість йоду, який відтитровують розчином натрію тіосульфату. Індикатор – крохмаль, який також додають у кінці титрування.
Йодометричний метод застосовують також для визначення водовмісних органічних сполук після переведення йоду в іоноген ний стан, окисленням до йодату.Цериметрія. Метод заснований на застосуванні в якості окислювача розчинів солей церію (IV), який в кислих розчинах – сильний окислювач: приєднуючи електрон, він відновлюється до церію (ІІІ), причому реакція ця зворотня.
22. Потенціометричне титрування, як і кондуктометричне, використовують у разі визначення концентрації забарвлених або каламутних розчинів, де неможливе застосування кольорових індикаторів, а також для визначення вмісту окремих компонентів у сумішах кислот або основ, константи дисоціації яких суттєво відрізняються, і у сумішах електролітів, які внаслідок додавання титранту утворюють осади з суттєво різними добутками розчинності.
Для визначення концентрації досліджуваного розчину складають гальванічний елемент (ГЕ) з двох електродів: електрода порівняння, потенціал якого залишається сталим у процесі титрування, та індикаторного електрода, потенціал якого залежить від концентрації (активності) іонів, що входять до складу утвореного внаслідок додавання титранту осаду або слабкого електроліту. Еквівалентний об’єм визначають за графічними залежностями  (інтегральна крива) ). Поблизу точки еквівалентності потенціал індикаторного електрода різко змінюється, що призводить у свою чергу до стрибка електрорушійної сили (ЕРС) ГЕ. Стрибки потенціалу індикаторного електрода і ЕРС ГЕ будуть тим більші, чим менші розчинність або ступінь дисоціації утвореної внаслідок додавання титранту сполуки. Еквівалентний об’єм
(інтегральна крива) ). Поблизу точки еквівалентності потенціал індикаторного електрода різко змінюється, що призводить у свою чергу до стрибка електрорушійної сили (ЕРС) ГЕ. Стрибки потенціалу індикаторного електрода і ЕРС ГЕ будуть тим більші, чим менші розчинність або ступінь дисоціації утвореної внаслідок додавання титранту сполуки. Еквівалентний об’єм  визначають за точкою максимального перегину інтегральної кривої..
визначають за точкою максимального перегину інтегральної кривої..
Потенціометричне титрування має низку переваг перед індикаторними титриметричними методами: об’єктивність і точність встановлення к.т.т., невисока нижня межа концентрацій, які визначаються, можливість титрування каламутних та забарвлених розчинів, можливість диференційованого (кожного окремо) визначення компонентів із однієї аліквоти аналізованого розчину, якщо відповідні φ0 достатньо різняться (Δ φ0 >0,2 В). Потенціометричне титрування можна виконувати автоматично до заздалегідь заданого значення різниці потенціалів, криві титрування (потенціограми) записують як в інтегральній, так і в диференціальній формі. За потенціограмами можна визначати удавані константи рівноваги (напр. змішані константи дисоціації кислот і основ) та значення потенціалу середньої точки (мідпойнт-потенціалу) φm (коли аох=аred).
Для визначення компонентів оборотних систем, коли на електродах встановлюються рівноважні значення потенціалів, потенціометричне титрування здійснюють при силі струму І=0. У випадку необоротних електродних процесів перевагу надають титруванню досліджуваного розчину з одним або двома поляризованими електродами, тобто при контрольованій силі струму. Потенціометричні методи аналізу широко використовують для автоматизації контролю технологічних процесів у хімічній, фармацевтичній, нафтопереробній, харчовій та інших галузях промисловості, в медицині, біології, геології, а також для проведення контролю забруднення довкілля.
23. Мембранні електричні потенціали існують фактично у всіх клітин організму. Деякі клітини, наприклад нервові і м'язові, здатні генерувати швидко-змінюються електрохімічні імпульси, які використовуються для передачі сигналів вздовж мембран цих клітин.У клітинах інших типів, наприклад залізистих, макрофагах і реснитчатих, локальні зміни мембранних потенціалів також активують багато клітинні функції. У цьому розділі обговорюються мембранні потенціали, що генеруються нервовими і м'язовими клітинами в спокої і в активному стані.
Дифузійний потенціал, Обумовлений розходженням іонних концентрацій по обидва боки мембрани. Концентрація іонів калію всередині нервового волокна - висока, але зовні -дуже низька. Припустимо, що в цьому випадку мембрана проникна для іонів калію, але непроникна для інших іонів. Через велику градієнта концентрації існує потужна тенденція до дифузії з клітки через мембрану великого числа іонів калію. В процесі дифузії вони виносять назовні позитивні електричні заряди, в результаті мембрана зовні заряджається позитивно, а всередині - негативно, оскільки залишилися всередині негативні аніони не дифундують з клітки разом з іонами калію.
ІОНОСЕЛЕКТИВНІ ЕЛЕКТРОДИ(мембранні електроди) — електрохімічні напівелементи електрохімічного ланцюга, потенціали яких залежать від активності у розчині іона, до якого селективний цей електрод. Потенціал ідеального І.е. описується рівнянням Нернста:

де Е0 — стандартний електродний потенціал електрода; R — газова постійна; T — температура розчину, К; n — заряд іона з відповідним знаком; ai — активність іона, до якого селективний електрод.
Розроблені і знайшли застосування спеціальні І.е., напр., газочутливі для потенціометричного визначення деяких газів: NH3, CO2, SO2, H2S; ферментні для визначення неіоногенних органічних сполук: сечовини, глюкози та ін.; бактеріальні, імуноелектроди, електроди на основі біологічної тканини таін.
І.е. широко застосовують у фармації, загальнохімічному аналізі, у дослідженні механізмів хімічних реакцій, у медико-біологічних дослідженнях, контролі навколишнього середовища тощо
1. ВМС називаються такі речовини, що мають молекулярну масу від декількох тисяч до мільйона і більше.
Молекули цих сполук з такою великою молекулярною масою (не нижче 10–15 тис.) складаються із сотень і навіть тисяч окремих атомів, зв'язаних один з одним силами головних валентностей.
Кожна молекула ВМС представляє гігантське утворення, тому такі молекули прийнято називати макромолекулами.
Велика молекулярна маса обумовлює і великі розміри молекул з мікродіаметром у поперечнику.
Характерною рисою більшості ВМС є наявність у їхніх молекулах багаторазово повторюваних ланок. Це повторення залежить від ступеня полімеризації. Звідси ці речовини мають ще і другу назву– полімери.
Властивості ВМС залежать не тільки від величини молекул, але і від форми молекул. Молекули полімерів бувають лінійні, сферичні, площинні, тривимірні. Так, наприклад, ВМС володіючі сферичними молекулами (глікоген, гемоглобін), при розчиненні майже не набухають. Це зумовлено тим, що сили зчеплення між молекулами завжди менші сил, що приходяться на ланцюгову молекулу. Тому розчинення таких речовин відбувається легше.
ВМС із сильно асиметричними лінійними витягнутими молекулами (желатин, целюлоза, її ефіри, каучук і ін.) при розчиненні дуже сильно набухають і утворять грузлі розчини.
Гігантські ланцюговоподібні молекули ВМС по окремих ланках неоднорідні, мають дифільний характер. Окремі ланки складаються з атомних груп, що мають полярний характер. До числа полярних груп належать –СООН, -NН2, -ВІН, і ін. Ці радикали добре взаємодіють з полярними рідинами (водою, спиртом і ін.) – гідратуються інакше кажучи, вони гідрофільні. Поряд з полярними макромолекула може містити неполярні гідрофобні радикали: -СН3, -СН2, -З6Н5 і ін., що можуть сольватуватися неполярними рідинами (бензол, петролейний ефір і т.п.), але не можуть гідратуватися.
У природних ВМС майже завжди переважають полярні групи, тому, потрапляючи у воду, вони поводяться як гідрофільні речовини: чим більше полярних ділянок у молекулі ВМС, тим краще вони розчинні у воді.
Завдяки цим властивостям ВМС, особливо білки, широко застосовуються як стабілізатори при виготовленні деяких фармацевтичних препаратів і ін'єкційних форм. Причина стабілізуючої дії ВМС полягає в тім, що вони адсорбуються на частках гідрофобного колоїду й у результаті чого гідрофобна речовина здобуває характер гідрофільної колоїдної системи
Однобічний процес проникнення молекул розчинника у фазу полімеру називається набуханням. Залежно від будови макроланцюга і характеру взаємодії макромолекул між собою і молекулами розчинника розрізняють обмежене і необмежене набухання. Необмежене набухання — це набухання, яке спонтанно переходить у розчинення, при якому утворюється однофазна гомогенна система. Обмеженим набуханням називається процес взаємодії полімеру з низькомолекулярною рідиною, обмежений стадією набухання. Спонтанне розчинення полімеру не відбувається, тобто його ланцюги повністю не відділяються один від одного. При цьому утворюються дві співіснуючі фази. Одна фаза є розчином низькомолекулярної рідини в полімері, а друга — чистою низькомолекулярною рідиною. Ці фази розділені видимою поверхнею поділу і перебувають у рівновазі. Якщо в полімері є просторова сітка, утворена хімічними зв’язками, то ланцюги макромолекул ні за яких температур не можуть бути розділені. Отже, просторові полімери принципово нерозчинні, однак вони можуть набухати, утворюючи драглі або гелі. Процес набухання кількісно характеризується ступенемі швидкістюнабухання.Ступіньнабухання(a) виражається кількістю рідини, поглиненої одиницею маси або об’єму полімеру На здатність полімерів утворювати гомогенні системи з низькомолекулярними речовинами впливають різні фактори. Одним із них є природа полімеру і розчинника. ВМС з ізодіаметричними молекулами (напр. гемоглобін, глікоген) при розчиненні майже не набухають, а розчини цих речовин мають невисоку в’язкість навіть при дуже високих концентраціях від ступеня полярності полімеру й розчинника. Якщо ланки ланцюгів і молекули розчинника близькі за полярністю, то настає набухання й розчинення полімеру. Якщо
|
Просмотров 591 |
|
|
