Главная Обратная связь
Дисциплины:
Архитектура (936)
Биология (6393)
География (744)
История (25)
Компьютеры (1497)
Кулинария (2184)
Культура (3938)
Литература (5778)
Математика (5918)
Медицина (9278)
Механика (2776)
Образование (13883)
Политика (26404)
Правоведение (321)
Психология (56518)
Религия (1833)
Социология (23400)
Спорт (2350)
Строительство (17942)
Технология (5741)
Транспорт (14634)
Физика (1043)
Философия (440)
Финансы (17336)
Химия (4931)
Экология (6055)
Экономика (9200)
Электроника (7621)

АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ (ЦИКЛОАЛКАНЫ)
|
|
ГЛАВА 44.
ТЕРПЕНЫ
Краткая история исследования химии терпенов
Терпенами называют углеводороды и их кислородсодержащие производные, входящие в состав эфирных масел и смол хвойных и других растений. Химическая структура различных терпенов имеет много общего. Их молекулы включают разное число связанных между собой остатков изопрена:

Поэтому общая суммарная формула всех терпенов является кратной от C5H8, т.е. (C5H8)n. Терпены могут иметь ациклическую и циклическую структуру. Среди терпенов различают монотерпены C10H16, сесквитерпены C15H24, дитерпены C20H32, тритерпены C30H48 и политерпены (C5H8)n. Циклические терпены могут иметь моно- и бициклическую структуру.
Первые исследования в области синтеза и изучения химической структуры терпенов относятся к началу XIX в. В 1803 г. аптекарь Кинд получил борнилхлорид («искусственную камфору») из скипидара. Д. И. Менделеев исследовал эфирные масла и смолы. Большое влияние на изучение химии терпенов оказали проведенные А.М. Бутлеровым исследования отечественных эфирных масел. В начале XX в. Е.Е. Вагнером была установлена структура входящих в состав сосновых скипидаров пинена, камфена, лимонена и других терпенов. Изучению биосинтеза терпенов, в частности сесквитерпенов, посвятил многие свои работы Л. С. Ружичка. Крупные исследования в области терпенов выполнили русские ученые А.М. Зайцев и Ф.М. Флавицкий, исследовавшие состав эфирных масел различных пород деревьев, С.Н. Реформатский и В.В. Марковников, подробно изучившие состав розового масла. В познание химии и стереохимии терпенов большой вклад внесли труды Московской и С.-Петербургской школ академиков H.Д. Зелинского, С.С. Наметкина, А.Е. Фаворского, В.Е. Тищенко. Особенно значительны высказанные ими теоретические положения о путях превращений многих би- и трициклических терпенов (С.С. Наметкин и др.) и данные об оптической активности терпенов (Л.А. Чугаев). Ф. М. Флавицкий впервые предложил классификацию терпенов, осуществил реакции взаимопревращения моноциклических терпенов в бициклические. Он явился родоначальником Казанской школы в области химии терпенов. Его преемниками стали А. Е. Арбузов, разработавший новую совершенную технику сбора живицы, и Б. А. Арбузов, открывший изомеризацию терпенов.
Большой интерес крупнейших русских химиков к исследованию терпенов объясняется прежде всего их важным народнохозяйственным значением. Не случайно поэтому теоретические исследования в области расшифровки химической структуры этой группы веществ тесно переплетались с разработкой способов синтеза камфоры из скипидара (В.Е. Тищенко, Т.А. Рудаков, П.П. Шорыгин) и пихтового масла (П.Г. Голубев, Н.В. Вершинин), установлением состава канифоли (Ф.М. Флавицкий, Б.А. Арбузов, В.В. Крестинский), получения скипидара и канифоли из отходов сосновых деревьев (В.М. Руднев).
Лекарственные вещества из класса терпенов классифицируют (по количеству циклов) на моноциклические терпены и бициклические терпены. Их производные — терпеноиды — по характеру функциональных групп разделяют на спирты, альдегиды, кетоны, сложные эфиры, кислоты и т.д.
Моноциклические терпены
Три лекарственных вещества из числа моноциклических терпенов: ментол, валидол и терпингидрат по химическому строению представляют собой производные гидроароматического углеводорода — ментана:

Ментол получают из мятного масла, содержащегося в различных видах мяты, и синтетическим путем. Мятное масло обычно содержит от 40 до 80% ментола или ментилового эфира уксусной кислоты. Для переработки мятного масла с высоким содержанием ментола (до 80%) используют способ вымораживания. Он основан на фракционной перегонке масла, выделении фракции, кипящей при 208–212 °C (содержащей ментол), и охлаждении этой фракции до –16 — –20 °C. Выделившиеся кристаллы ментола отжимают и перекристаллизовывают.
Для сортов масла, содержащих 50–60% ментола, используют боратный способ. Мятное масло нагревают с борной кислотой:

Полученный ментиловый эфир борной кислоты имеет высокую температуру кипения, что позволяет отделить его от других компонентов мятного масла. Затем эфир омыляют и получают ментол. Процесс омыления легко проходит при перегонке с водяным паром.
Ментол синтезируют из природного тимола или реакцией алкилирования из м-крезола (до тимола) с последующим гидрированием:

Валидол представляет собой не индивидуальное вещество, а 25%-ный раствор ментола в ментиловом эфире изовалериановой кислоты. Последний синтезируют с помощью реакции этерификации:

Затем в полученном эфире растворяют ментол.
В качестве исходного продукта для получения терпингидрата используют скипидар, основным компонентом которого является пинен. Скипидар подвергают фракционной перегонке и выделяют кипящую при 155–161°C фракцию пинена. Пинен затем подвергают гидратации. Процесс этот протекает медленно (10–14 дней), на холоду. Для достижения хорошего контакта в промышленных условиях пинен смешивают с древесными опилками и заливают 25–30%-ным раствором серной кислоты. Затем смесь нейтрализуют содой, отделяют терпингидрат, очищают его и перекристаллизовывают:

По физическим свойствам ментол и терпингидрат — бесцветные кристаллические вещества, а валидол — жидкость (табл. 44.1). Ментол и валидол отличаются от терпингидрата характерным запахом мяты. В медицинской практике применяют l-ментол (левовращающий изомер ментола) и ментол рацемический, содержащий сумму стереоизомеров ментола, в т.ч. не менее 70% d,l-ментола . Ментол (l-изомер и рацемат) летуч при обычной температуре. Он образует эвтектические жидкие смеси при растирании с равными количествами камфоры, фенола, тимола, резорцина, хлоралгидрата.
41.1. Свойства моноциклических терпенов
| Лекарственное вещество | Химическая структура | Описание |
| Mentholum— ментол |  l-2-изопропил-5-метилциклогексанол-1
l-2-изопропил-5-метилциклогексанол-1
| Бесцветные кристаллы с сильным запахом перечной мяты и холодящим вкусом. Т. пл. 41–44°C. Удельное вращение от -49 до –51° (10%-ный раствор в этаноле) |
| Mentholum racemicum— ментол рацемический (d,l-ментол) |  d, l-2-изопропил-5-метилциклогексанол-1
d, l-2-изопропил-5-метилциклогексанол-1
| Бесцветные кристаллы или твёрдая кристаллическая масса с сильным запахом перечной мяты и холодящим вкусом. Т. пл. 28-32°C. Удельное вращение от –0,5 до +0,5 ° (10%-ный раствор в этаноле) |
| Validolum— валидол |  раствор ментола в ментиловом эфире
изовалериановой кислоты (или в смеси ментиловых эфиров изовалериановой и метилэтилуксусной кислот)
раствор ментола в ментиловом эфире
изовалериановой кислоты (или в смеси ментиловых эфиров изовалериановой и метилэтилуксусной кислот)
| Прозрачная маслянистая бесцветная или слегка окрашенная жидкость с запахом ментола. Пл. 0,894–0,907 г/см3. Показатель преломления от 1,4490 до 1,4515 |
| Terpinum hydratum— терпингидрат |  n-ментандиол-1,8
n-ментандиол-1,8
| Бесцветные прозрачные кристаллы или белый кристаллический порошок без запаха. Т. пл. 115–117°C |
Терпингидрат возгоняется при медленном нагревании до 100°C, образуя при последующем охлаждении игольчатые кристаллы. При быстром нагревании до 115–117°C терпингидрат плавится, теряя молекулу воды и превращаясь в цис-терпин (белую кристаллическую массу с т. пл. 102–103°C).
Ментол и терпингидрат характеризуются очень малой растворимостью в воде, валидол практически нерастворим в воде. В этаноле ментол и валидол очень легко растворимы, а терпингидрат растворим. Ментол в отличие от терпингидрата очень легко растворим в эфире, жирных маслах, вазелиновом масле.
Подлинность ментола рацемического подтверждают по ИК-спектру, снятому в 10%-ном растворе тетрахлорметана в области 3600-870 см–1 и в парафиновом масле в области 870-750 см–1. Он должен полностью совпадать с полосами поглощения прилагаемого к ФС рисунка спектра.
Для идентификации ментола и валидола ФС рекомендуют цветную реакцию с концентрированной серной кислотой в присутствии ванилина. Наблюдается появление желтого окрашивания, которое при добавлении воды переходит в малиново-красное (тимол этой реакции не дает). Реакция основана на окислении и взаимодействии активированной метиленовой группы ментола с ароматическим альдегидом:

Ментол дает цветную реакцию с о-фталевым ангидридом; в присутствии концентрированной серной кислоты появляется оранжево-красное окрашивание. Спиртовой раствор фурфурола после смешения с ментолом и концентрированной серной кислотой приобретает фиолетовую окраску. В присутствии ментола наблюдается также изменение окраски бензольного раствора оксихинолината ванадия. Серо-зеленая окраска реактива при нагревании на водяной бане переходит в красную.
Терпингидрат после добавления концентрированной серной кислоты образует мутный раствор и приобретает характерный запах, обусловленный образованием a-терпинеола, цинеола (эвкалиптола) и других продуктов дегидратации:

Терпингидрат образует окрашенные продукты после выпаривания его смеси со спиртовым раствором хлорида железа (III). Если растворить остаток в бензоле, он окрашивается в синий цвет.
Примесь тимола (источник синтеза) в ментоле устанавливают реакцией со смесью концентрированной серной и азотной кислот. Определяют также наличие примесей легко окисляющихся веществ по реакции с раствором перманганата калия и нелетучий остаток (после нагревания до 100-105 °C), который не должен превышать 0,05%. В валидоле после выпаривания на водяной бане досуха остаток не должен превышать 0,1%. Ментол и валидол испытывают на микробиологическую чистоту, кислотность, прозрачность и цветность спиртовых растворов.
Количественное определение ментола ФС рекомендует выполнять двумя методами — химическим и ГЖХ.
Определение суммы стереоизомеров ментола основано на ацетилировании уксусным ангидридом в среде безводного пиридина (при нагревании с обратным холодильником). Избыток уксусного ангидрида разлагают водой до уксусной кислоты и титруют ее 0,5 М раствором гидроксида натрия (индикатор фенолфталеин):

Параллельно проводят контрольный опыт. Общая сумма стереоизомеров ментола должна быть не менее 99%.
Альтернативным методом, рекомендуемым ФС для количественного определения ментола рацемического, является ГЖХ. Причём одновременно определяют примеси изо-d,l-ментола и нео-d,l-ментола. Массовая доля этих примесей должна быть не более 30%. Расчёты выполняют по площадям пиков.
Ментол можно определить термонефелометрическим методом, а также фотоколориметрически на основе цветных реакций с альдегидами. Оптимальным реактивом является 1%-ный раствор n-диметиламинобензальдегида в концентрированной серной кислоте, а растворителем — смесь этанола с водой (1:4). Фотоколориметрическое определение выполняют при 545 нм. В этих же условиях определяют компоненты, входящие в состав валидола, а также терпингидрат (при 490 нм) в таблетках.
В валидоле количественно определяют содержание ментилового эфира изовалериановой кислоты, омыляя его 0,5 М спиртовым раствором гидроксида калия (кипятят 5 ч с обратным холодильником):

Избыток гидроксида калия оттитровывают 0,5 М раствором хлороводородной кислоты (индикатор фенолфталеин).
Проведенными исследованиями методом ГЖХ (Н.С. Евтушенко) установлено, что валидол представляет собой более сложную по составу смесь, содержащую кроме 22,2–25,7% ментола и 52,7–55,7% ментилового эфира изовалериановой кислоты, также 17,4–18,9% ментилового эфира 2-метилмасляной кислоты и 2,2–5,9% углеводородов ментенового ряда:

С использованием метода внутреннего стандарта разработаны методики одновременного испытания на подлинность, чистоту и количественного определения каждого из компонентов, входящих в состав валидола. Методики включены в ФС на валидол. В валидоле при испытании подлинности устанавливают методом ГЖХ наличие всех указанных компонентов, сравнивая относительные времена удерживания валидола и модельной смеси. Относительное время удерживания ментола принимают за единицу.
При испытании на чистоту валидола общее содержание примесей не должно быть более 6%. Допускается присутствие ментена-1, ментена-2, ментена-3, а также других неидентифицированных примесей. Их содержание определяют по сумме площадей пиков всех примесей по отношению к сумме площадей пиков всех компонентов. При количественном определении таким же образом рассчитывают содержание ментола (17–28%) и суммы ментиловых эфиров изовалериановой и метилэтилуксусной кислот (68,5–75%).
Для определения терпингидрата в таблетках рекомендован гравиметрический метод, основанный на извлечении его этанолом, удалении последнего при нагревании на водяной бане, высушивании остатка до постоянной массы в эксикаторе и взвешивании.
Лекарственные препараты моноциклических терпенов хранят в хорошо укупоренной таре в прохладном месте, так как ментол и валидол летучи даже при комнатной температуре, а терпингидрат в сухом теплом воздухе медленно выветривается, теряя молекулу кристаллизационной воды. Ментол при хранении следует защищать от действия света и влаги. Температура воздуха при хранении не должна превышать +15 °C.
Ментол применяют наружно как успокаивающее, болеутоляющее и слабое антисептическое средство при воспалительных заболеваниях верхних дыхательных путей в виде 0,5–5%-ных спиртовых и масляных растворов. Ментол применяют внутрь (1–2 капли 5%-ного спиртового раствора на сахаре для подъязычного применения) при стенокардии как спазмолитическое средство. Однако чаще для этого используют валидол по 4–5 капель на сахар или в виде капсул, таблеток (по 0,06 г). Терпингидрат применяют внутрь как отхаркивающее средство по 0,25–0,3 г при хронических бронхитах.
Бициклические терпены
В качестве лекарственных веществ используют природный бициклический терпен — камфору и ее производные — бромкамфору и кислоту сульфокамфорную. Эти вещества представляют собой производные углеводорода камфана (борнилана). Камфора является кетопроизводным камфана:

Ввиду наличия в молекуле двух асимметрических атомов углерода существуют d-камфора (правовращающий изомер), l-камфора (левовращающий изомер) и рацемическая камфора.
Природную d-камфору (японскую камфору) получают из камфорного дерева, произрастающего в Японии и в Китае (в основном на острове Тайвань). Получение сводится к отгонке камфоры водяным паром из измельченной древесины. Затем камфору подвергают очистке возгонкой и отжимают на прессах.
Поскольку потребность в камфоре не удовлетворяется природными источниками, были разработаны синтетические и полусинтетические способы ее получения. В нашей стране способ получения камфоры был впервые разработан П.Г. Голубевым (Военно-медицинская академия). В 1934 г. в Новосибирске на опытном заводе было организовано промышленное производство синтетической камфоры.
Исходным продуктом является пихтовое масло, которое перегоняют с водяным паром из «пихтовых лапок» (концов веток пихт). Пихтовое масло состоит из борнилацетата (30–40%), камфена (10–20%), пинена (10%) и других веществ. Фракционной перегонкой при температуре выше 180°C выделяют фракцию пихтового масла, содержащую борнилацетат. Борнилацетат подвергают омылению с помощью гидроксида натрия, а затем окислению (хромовой смесью или азотной кислотой) до образования камфоры:

Из пихтового масла получается синтетическая l-камфора. Благодаря исследованиям, проведенным Н.А. Вершининым, доказана идентичность ее по терапевтическому действию японской d-камфоре.
Синтетическую камфору (рацемическую или d,l-форму) можно получить способом, предложенным В.Е. Тищенко, из пинена, содержащегося в скипидаре. Пиненовую фракцию скипидара изомеризуют в камфен с помощью катализатора, содержащего оксид титана (IV). Затем при взаимодействии с муравьиной кислотой получают борнилформиат (рацемическую форму). Последующий процесс получения камфоры идентичен способу, предложенному П.Г. Голубевым. Общая схема синтеза:
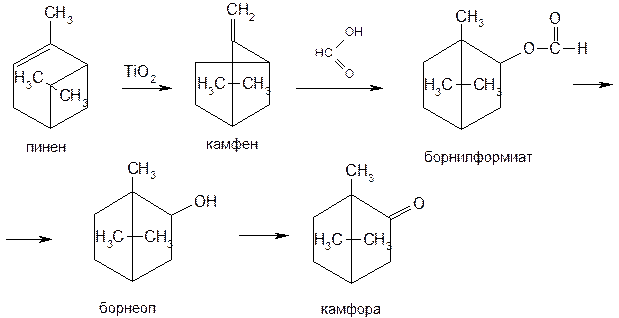
Бромкамфору получают действием брома на d-камфору. Реакцию выполняют в среде хлороформа или хлоралгидрата. Растворитель отгоняют, а бромкамфору перекристаллизовывают:

По этой же схеме получают бромкамфору из l-камфоры и рацемической камфоры. Исследования в области создания и стандартизации лекарственных средств на основе бромкамфоры рацемической были выполнены в Уральском НИИ технологии медицинских препаратов и Пятигорской фармацевтической академии.
По физическим свойствам камфора, бромкамфора и кислота сульфокамфорная отличаются друг от друга (табл. 44.2). Это используется для подтверждения их подлинности.
44.2. Свойства бициклических терпенов
| Лекарственное вещество | Химическая структура | Описание |
| Camphora— камфора |  1,7,7-триметилбицикло[2.2.1]-гептан-2-он
1,7,7-триметилбицикло[2.2.1]-гептан-2-он
| Белые кристаллические куски или бесцветный кристаллический порошок, или прессованные плитки с кристаллическим строением, легко режущиеся ножом, слипающиеся в комки |
| Bromcamphora racemata— бромкамфора рацемическая |  d,l-3-бром-1,7,7-триметилбицикло[2.2.1]-гептан-2-он
d,l-3-бром-1,7,7-триметилбицикло[2.2.1]-гептан-2-он
| Бесцветные кристаллы или белый кристаллический порошок камфорного запаха. Т. пл. 48–53 °C. Удельное вращение от –1 до +1° (10%-ный раствор в этаноле) |
| Acidum sulfocamphoratum— кислота сульфокамфорная | 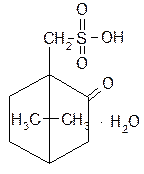 1,7,7-триметилбицикло[2.2.1]-гептан-2-он-сульфоновая-10-кислота, моногидрат
1,7,7-триметилбицикло[2.2.1]-гептан-2-он-сульфоновая-10-кислота, моногидрат
| Белый или со слегка желтоватым оттенком кристаллический порошок. Т. разложения 194–198 °C для левовращающей и 196-202 °C для рацемической. Удельное вращение от –20° до –24° для левовращающей и от –1 до +1° для рацемической (5%-ный водный раствор) |
Камфора отличается сильным характерным запахом и пряным горьковатым, а затем охлаждающим вкусом. У бромкамфоры камфорный запах и вкус менее выражены. Растворимость бициклических терпенов находится в зависимости от химической структуры, т.е. наличия в молекуле тех или иных функциональных групп. Камфора и бромкамфора практически нерастворимы в воде, легко растворимы в этаноле, очень легко в эфире, хлороформе, легко растворимы в жирных маслах. Кислота сульфокамфорная, содержащая в молекуле сульфогруппу, очень легко растворима в воде и этаноле, но мало растворима в эфире.
Подобно ментолу, камфора образует густые прозрачные жидкости (эвтектические смеси) с фенолом, ментолом, тимолом, хлоралгидратом, а также постепенно возгоняется даже при обычной температуре, образуя в верхних частях сосуда кристаллический сублимат. При осторожном нагревании камфора полностью возгоняется без обугливания. Горит светлым пламенем, флуоресцирует в УФ-свете.
Утверждённая в 1999 г ФС распространяется на l-камфору, получаемую из пихтового масла, и рацемическую — получаемую из скипидара. Они отличаются друг от друга некоторыми константами (табл. 44.3).
44.3. Физические константы камфоры (по ФС)
| Лекарственное вещество | Темп. затвердевания, °C | Удельное вращение (10%-ный раствор в этаноле) | Темп плавл. 2,4-динитрофенилгидразона, °C | Темп. кипения (возгон.), °C |
| d-камфора | 178,2-178,6 | +44,3° | — | 207,4-209,1 |
| l-камфора | 174-179 | от –39 до -44° | 174-176 | —«— |
| Камфора рацемическая | 171-177 | от 1,0 до + 1,0° | 164-167 | —«— |
Эти константы служат подтверждением подлинности камфоры. Однако для ее идентификации могут быть также использованы цветные реакции, основанные на взаимодействии активированной метиленовой группы с альдегидами: фурфуролом (сине-фиолетовое окрашивание), бензальдегидом (красное):

Наличие в молекулах камфоры, бромкамфоры и кислоты сульфокамфорной кетогруппы обусловливает ряд других химических реакций, которые используют для их испытания подлинности и количественного определения. C этой целью применяют реакции образования оксимов, фенилгидразонов, семикарбазонов. ФС рекомендует для подтверждения подлинности камфоры реакцию образования 2,4-динитрофенилгидразона и установления его температуры плавления (табл. 44.3). Раствор (0,006%) 2,4-динитрофенилгидразона камфоры в этаноле в области 220–450 нм имеет максимумы поглощения при 231, 365 нм и плечо от 255 до 275 нм. Подлинность камфоры можно подтвердить по температурам плавления и УФ-спектрам других производных камфоры.
Известны способы идентификации спиртовых растворов камфоры и бромкамфоры с использованием метода УФ-спектрофотометрии. Камфора имеет максимум светопоглощения при 231 и 365 нм, плечо — от 273 до 277 нм с незначительным удельным показателем поглощения (около 2), бромкамфора при 306 нм (удельный показатель поглощения 4,34).
Подлинность бромкамфоры устанавливают по идентичности ИК-спектра испытуемого вещества в области от 4000 до 400 см–1 и рисунка спектра, прилагаемого к ФС. Этот метод используют также для количественного определения.
При нагревании бромкамфоры с концентрированной серной кислотой раствор приобретает красно-бурое окрашивание. После охлаждения и осторожного добавления воды жидкость сохраняет красно-бурый цвет.
Испытание на подлинность и количественное определение бромкамфоры (по ФС) основано на отщеплении атома брома от органической части молекулы. Этот процесс происходит в сравнительно «мягких» условиях при нагревании бромкамфоры в присутствии цинковой пыли и гидроксида натрия в течение 1–2 мин:

В фильтрате затем обнаруживают бромид-ионы по реакции с хлорамином (см. ч. 2, гл. 21).
Для количественного определения навеску бромкамфоры кипятят в присутствии 30%-ного раствора гидроксида калия и цинковой пыли в течение 30 мин, после чего используют обратное аргентометрическое титрование для определения образовавшегося эквивалентного количества бромида калия.
Устанавливают содержание бромкамфоры и методом сжигания в кислороде. Поглотительным раствором служит раствор пероксида водорода. Образовавшийся бромид-ион определяют меркуриметрическим методом. В лекарственных формах бромкамфору определяют рефрактометрическим и спектрофотометрическим методом при 306-308 нм (растворитель этанол).
Утверждённая ФС регламентирует требования на кислоту сульфокамфорную, получаемую из l-камфоры и рацемической камфоры. В зависимости от способа получения кислота сульфокамфорная отличается по температуре разложения и удельному вращению (табл. 44.2).
При испытании подлинности кислоты сульфокамфорной подтверждают наличие в её молекуле сульфо- и кетогрупп. Присутствие сульфогруппы можно установить, разрушая лекарственное вещество при прокаливании его в смеси карбоната и нитрата натрия с последующим воздействием концентрированной хлороводородной кислотой. Образовавшийся сульфат-ион обнаруживают по реакции с раствором хлорида бария. Наличие кетогруппы подтверждают по образованию желто-оранжевого осадка при взаимодействии с раствором 2,4-динитрофенилгидразина:

При испытании бициклических терпенов на чистоту ФС предусматривают установление прозрачности, цветности, pH растворов, микробиологической чистоты. В кислоте сульфокамфорной определяют наличие примесей ацетатов, сульфатов, потерю в массе при высушивании. Камфору контролируют на содержание воды, жирных масел, нелетучего остатка. Наличие в бромкамфоре посторонних веществ определяют методом ГЖХ, устанавливая по площадям пиков присутствие дибромкамфоры и неидентифицированных примесей (не более 1%). Столь высокие требования к чистоте обусловлены особенностями физических свойств и применения указанных лекарственных веществ.
Для количественного определения камфоры используют оксимный способ, основанный на взаимодействии камфоры с определенным количеством гидрохлорида гидроксиламина:
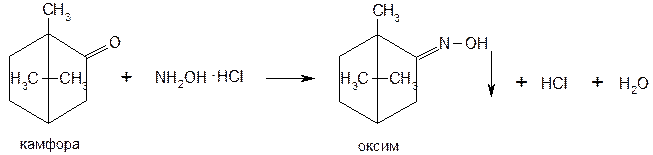
Нерастворимый в воде оксим определяют гравиметрическим методом или титруют выделившееся эквивалентное количество хлороводородной кислоты титрованным раствором гидроксида натрия. Количественное определение камфоры может быть также выполнено гравиметрическим методом по содержанию 2,4-динитрофенилгидразона, полученного при нагревании навески в течение 4 ч (с обратным холодильником) с 2,4-динитрофенилгидразином.
Испытание на подлинность и количественное определение камфоры согласно ФС выполняют методом ГЖХ с помощью хроматографа, снабженного детектором по теплопроводности или детектором по ионизации в пламени. Подлинность подтверждают, снимая хроматограммы двух ацетоновых растворов. Один из них содержит только испытуемое вещество, а в другой прибавляют синтетической левовращающей или рацемической камфоры. О подлинности судят по значительному увеличению основного пика камфоры. Количественное определение выполняют методом внутренней нормализации в тех же условиях. Содержание камфоры вычисляют по отношению площади пика камфоры к сумме площадей всех пиков компонентов лекарственного вещества на хроматограмме. Оно должно быть не менее 97% для камфоры, применяемой для инъекций, и не менее 94% для камфоры, используемой для наружного применения. Одновременно по относительным временам удерживания устанавливают возможные примеси трициклена, камфена, фенхона, изофенхона, изофенхола, борнеола, изоборнеола и др.
Кислоту сульфокамфорную количественно определяют методом нейтрализации в водной среде (индикатор фенолфталеин):
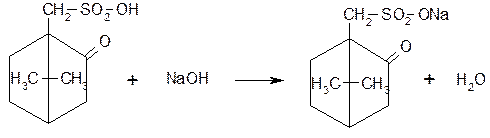
Лекарственные препараты бициклических терпенов хранят в хорошо укупоренных банках. Камфора должна находиться в прохладном месте, учитывая ее способность возгоняться. Бромкамфору хранят по списку Б при температуре не выше 25 °C, в банках оранжевого стекла в защищенном от света месте, чтобы избежать разложения с выделением брома. Кислоту сульфокамфорную хранят в сухом, защищенном от света месте.
Камфору применяют в качестве стимулятора центральной нервной системы и кардиотонического средства. Назначают внутрь (0,1–0,2 г) или подкожно в виде 20%-ного масляного раствора. При наружном применении камфора оказывает местное раздражающее и антисептическое действие. Бромкамфору применяют внутрь по 0,1–0,5 г как средство, успокаивающее центральную нервную систему. Специфическое аналептическое действие камфоры обусловлено наличием в ее молекуле карбонильной группы, а также активируемой ею ближайшей метиленовой группы. Любое изменение структуры камфоры путем введения заместителей ослабляет ее аналептическую и кардиотоническую активность. Бром, введенный в молекулу, приводит к появлению седативного эффекта у бромкамфоры.
Кислота сульфокамфорная является составной частью применяемого для инъекций сульфокамфокаина (Sulfocamphocainum 10% pro injectionibus). Для его приготовления берут 49,6 г кислоты сульфокамфорной, 50,4 г основания прокаина и до 1 л воды для инъекций.
Сульфокамфокаин — прозрачная слегка желтоватая жидкость. Его подлинность устанавливают, выполняя характерную для первичных ароматических аминов реакцию образования азокрасителя (прокаин), а также реакцию на сульфогруппу, основанную на минерализации, окислении её до сульфат-иона и обнаружении последнего с помощью хлорида бария (органически связанная сера). Выполняют также цветную реакцию с 2,4-динитрофенилгидразином (кислота сульфокамфорная). Затем нитритометрическим методом количественно определяют содержание прокаина—основания и алкалиметрическим методом — содержание кислоты сульфокамфорной.
Фармакологическое действие сульфокамфокаина аналогично камфоре, но в связи с хорошей растворимостью в воде он быстро всасывается.
ГЛАВА 45.
СТАТИНЫ
Первые сведения о статинах (вастатинах) появились в 1987 г. Это принципиально новая группа гиполипидемических лекарственных веществ. Они блокируют функцию фермента 3-гидрокси-3-метил-глутарил-кофермента А-редуктазы (ГМГ-КоА-редуктазы). Последний катализирует начальные и промежуточные стадии биосинтеза холестерина. Вначале были получены из продуктов метаболизма грибов (рифомицетов), в частности, из штамма Aspergillus terreus: ловастатин и симвастатин, затем другие природные и несколько отличающиеся от них по химической структуре, синтетические аналоги статинов: правастатин, флувастатин и др.
Ловастатин и симвастатин являются пролекарствами, т. е. сами не обладают гиполипидемической активностью, но в организме метаболизируются до свободной b-оксикислоты, которая является конкурентным ингибитором ГМГ-КоА-редуктазы.
Общая формула статинов:

По химической структуре ловастатин и симвастатин очень сходны между собой. Молекула симвастатина в отличие от ловастатина включает дополнительно метильную группу в положении R1 (табл. 45.1).
45.1. Свойства ловастатина и симвастатина
| Лекарственное вещество | Химическая структура | Описание |
| Lovastatin— ловастатин (Мевакор) | 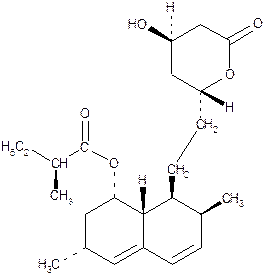
| Белый или почти белый кристаллический порошок. Удельное вращение от +325° до +340° (0,5%-ный раствор в ацетонитриле) |
| Simvastatin— симвастатин (Зокор) | 
| Белый или почти белый кристаллический порошок. Удельное вращение от +285° до +300° (0,5%-ный раствор в ацетонитриле) |
Ловастатин и симвастатин практически нерастворимы в воде. Ловастатин мало растворим в метаноле и этаноле, легко растворим в хлороформе, растворим в ацетоне. Симвастатин очень легко растворим в дихлорметане и легко растворим в этаноле.
ИК- и УФ-спектры ловастатина и симвастатина должны иметь полное совпадение с полосами поглощения спектра соответствующего стандартного образца. УФ-спектры спиртового раствора ловастатина имеют максимумы при 230, 238, 246 нм и минимумы — при 233 и 243 нм.
Методом ТСХ определяют посторонние примеси. Общее их содержание — не более 1%, каждой в отдельности — не более 3% от суммы примесей. Остаточные растворители определяют методом ГЖХ. Этилового спирта — не более 0,8%, изобутилацетата — не более 0,04%.
Количественно ловастатин и симвастатин определяют методом ВЭЖХ (от 98,5 до 101%). Для выполнения определения готовят для каждого по два рабочих раствора и по три стандартных раствора, используя в качестве растворителя ацетонитрил. В таблетках содержание ловастатина определяют спектрофотометрически в максимуме поглощения при 246 нм.
Хранят вастатины по списку Б в прохладном защищённом от света месте при температуре не выше 8 °C.
Применяют ловастатин и симвастатин для лечения гиперлипидемии и атеросклероза. Они снижают содержание общего холестерина в плазме крови и концентрацию липопротеидов низкой и очень низкой плотности, умеренно повышая концентрацию липопротеидов высокой плотности. Выпускают в виде таблеток: ловастатин по 0,1; 0,2 и 0,4 г; симвастатин по 0,01; 0,02 и 0,04 г, т.к. последний проявляет более высокую активность.
ГЛАВА 46.
ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА
|
Просмотров 11258 |
|
|
