Главная Обратная связь
Дисциплины:
Архитектура (936)
Биология (6393)
География (744)
История (25)
Компьютеры (1497)
Кулинария (2184)
Культура (3938)
Литература (5778)
Математика (5918)
Медицина (9278)
Механика (2776)
Образование (13883)
Политика (26404)
Правоведение (321)
Психология (56518)
Религия (1833)
Социология (23400)
Спорт (2350)
Строительство (17942)
Технология (5741)
Транспорт (14634)
Физика (1043)
Философия (440)
Финансы (17336)
Химия (4931)
Экология (6055)
Экономика (9200)
Электроника (7621)

Цвета растворов и соответствующих им светофильтров 2 часть
|
|
4. АНАЛИЗ ЛЕКАРСТВЕННЫХ ФОРМ
4.1. Дайте заключение о качестве раствора рибофлавина 0,02 % – 200 мл по количественному содержанию согласно приказу № 305.
Методика. К 0,5 мл лекарственной формы прибавляют 9,5 мл воды очищенной и измеряют оптическую плотность анализируемого раствора на фотоэлектроколориметре при длине волны 445 нм в кювете с толщиной поглощающего слоя 10 мм.
Параллельно измеряют оптическую плотность стандартного раствора, состоящего из 2,5 мл 0,004 % стандартного раствора рибофлавина и 7,5 мл воды очищенной.
В результате измерений на фотоэлектроколориметре оптическая плотность анализируемого раствора составляет 0,230, оптическая плотность стандартного раствора – 0,265.
Решение:
Содержание рибофлавина в граммах (Х) вычисляют по формуле:

,
где А, АГСО – оптические плотности испытуемого и стандартного растворов, соответственно;
СГСО – содержание рибофлавина в стандартном растворе с учётом разведения для измерения на фотоэлектроколориметре, г/мл;
V1 – объём лекарственной формы, взятой для анализа, мл,
V2 – объём разведения лекарственной формы для последующего измерения, мл;
V3 – общий объём лекарственной формы, мл.
 Концентрация стандартного раствора в г/мл ( СРСО) равна
Концентрация стандартного раствора в г/мл ( СРСО) равна
где 0,004 / 100 – переведение %-ной концентрации в г/мл;
2,5 – объём лекарственной формы, взятой для анализа, мл;
10(2,5+7,5) – объём разведения лекарственной формы, мл.
 Тогда:
Тогда:
Согласно приказу № 305 отклонения в массе рибофлавина массой свыше 0,02 до 0,1 (отклонения, допустимые в массе навески отдельных лекарственных веществ в жидких лекарственных формах при изготовлении массо- объёмным способом) должны быть не более ±15%
 В нашем случае отклонение составляет:
В нашем случае отклонение составляет:
Следовательно, лекарственная форма по количественному содержанию
рибофлавина приготовлена удовлетворительно.
4.2. Дайте заключение о качестве лекарственной формы состава:
Раствора рибофлавина 0,02 % – 10 мл;
Кислоты аскорбиновой 0,02
Тиамина бромида 0,02
Калия йодида 0,3
по количественному содержанию рибофлавина, если оптическая плотность раствора, полученного разведением 0,5 мл лекарственной формы до 10 мл водой, измеренная при длине волны 445 нм в кювете с толщиной поглощающего слоя 10 мм, равна 0,340. Удельный показатель поглощения раствора рибофлавина в максимуме при 445 нм равен 328.
4.3. Дайте заключение о качестве лекарственной формы состава:
Нитрофурала 0,2
Натрия хлорида 9,0
Воды для инъекций до 1 л
по количественному содержанию нитрофурала (фурацилина), если оптическая плотность раствора, полученного смешиванием 1 мл лекарственной формы, 15 мл воды и 4 мл 0,1 М раствора натрия гидроксида, измеренная при длине волны 450 нм в кювете с толщиной слоя 3 мм, равна 0,295. Оптическая плотность стандартного раствора, полученного из 1 мл 0,02 % раствора РСО нитрофурала по той же методике, равна 0,290. Содержание нитрофурала в 1 мл препарата должно быть не менее 0,000194 г и не более 0,000206 г.
4.4. Рассчитайте содержание хлорамфеникола (левомицетина) в лекарственной форме состава:
Раствора хлорамфеникола 0,015% 10 мл
Натрия хлорида 0,09 ,
если оптическая плотность 10 мл раствора, полученного из 1,5 мл разведения лекарственной формы 1:5, измеренная на фотоэлектроколориметре при длине волны 364 нм в кювете с толщиной слоя 5 мм, равна 0,430. Оптическая плотность 10 мл стандартного раствора хлорамфеникола, полученного из 1,5 мл 0,002 % раствора хлорамфеникола, измеренного в тех же условиях, равна 0,285.
4.5. При определении примеси свободной салициловой кислоты в таблетках кислоты ацетилсалициловой по 0,5 г точную навеску порошка растертых таблеток (0,5015 г) поместили в мерную колбу вместимостью 50 мл, прибавили 2 мл 0,2 % раствора железоаммониевых квасцов, довели спиртом до метки, профильтровали. Оптическая плотность раствора стандартного образца кислоты салициловой, полученного из 2 мл 0,01 % раствора в тех же условиях, равна 0,262. Средняя масса таблетки 0,605 г.
Сделайте заключение о качестве препарата по содержанию свободной салициловой кислоты, которой должно быть не более 0,000125 г, считая на среднюю массу одной таблетки.
4.6. Сделайте заключение о качестве таблеток нитроксолина 0,05 г, покрытых оболочкой, если при спектрофотометрическом определении точную массу порошка растертых таблеток (0,3975 г) поместили в мерную колбу вместимостью 250 мл, прибавили 20 мл воды и довели 0,2 М раствором натрия гидроксида до метки. После фильтрования 2 мл раствора разбавили 0,2 М раствором натрия гидроксида до 250 мл.
Оптическая плотность полученного раствора, измеренная при длине волны 450 нм в кювете с толщиной слоя 10 мм, составила 0,405.
Оптическая плотность раствора стандартного образца, содержащего 0,000003 г РСО нитроксолина в 1 мл, составила 0,395. Средняя масса одной таблетки 0,195 г. Содержание нитроксолина в одной таблетке должно быть не менее 0,04625 г и не более 0,05375 г.
ЛАБОРАТОРНЫЕ РАБОТЫ
Цель. Закрепление теоретических знаний и практических навыков работы на спектрофотометре и фотоэлектроколориметре. Использование спектрофотометрии как метода установления подлинности, чистоты, количественного содержания лекарственных веществ.
РАБОТА 1. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ПОДЛИННОСТИ И КОЛИЧЕСТВЕННОГО СОДЕРЖАНИЯ СУЛЬФАЦЕТАМИД НАТРИЯ
Практические навыки
1. Освоить технику измерения оптической плотности (светопропускания) на спектрофотометрах различных марок (СФ-46, СФ-56 и др.).
2. Приготовить растворы анализируемого и стандартного (ГСО) образцов сульфацетамид натрия (сульфацил-натрия).
3. Снять спектры приготовленных растворов и определить длины волн максимального и минимального поглощения.
4. Приготовить серию стандартных растворов сульфацетамид натрия, определить их оптические плотности и построить градуировочный график.
5. Доказать линейность зависимости A = f (с) в анализируемой области концентраций, рассчитать среднее значение удельного показателя поглощения (  ).
).
6. Определить количественное содержание сульфацетамид натрия, используя:
а) градуировочный график;
б) величину оптической плотности стандартного раствора, близкую к величине анализируемого раствора (см. Градуировочный график, рис.10, с.51);
в) численное значение удельного показателя поглощения ( ).
7. Написать отчет.
ВЫПОЛНЕНИЕ РАБОТЫ. Около 0,1г (точная масса) анализируемого
лекарственного вещества (сульфацетамид натрия) помещают в мерную колбу вместимостью 100 мл, растворяют в 50 мл воды и доводят объём до метки тем же растворителем, перемешивают (раствор А). 1 мл раствора А помещают в мерную колбу вместимостью 100 мл, доводят раствор водой до метки и перемешивают (раствор Б). Снимают УФ-спектры полученного раствора и раствора Государственного стандартного образца сульфацетамид натрия (сульфацил-натрия) в интервале длин волн от 220 до 340 нм.
Приготовление раствора стандартного образца (ГСО) сульфацетамид натрия 0,1 г (точная навеска) сульфацетамид натрия помещают в мерную колбу вместимостью 100 мл, растворяют в 50 мл воды и доводят объём до метки тем же растворителем, перемешивают (раствор А). 1 мл раствора А стандартного образца содержит 0,0010 г сульфацетамид натрия (сульфацил-натрия).
Построение градуировочного графика. Приготовить в мерных колбах вместимостью 100 мл серию стандартных растворов, состоящих из 0,3; 0,5; 0,7; 1; 1,3 мл 0,1 % раствора стандартного образца сульфацетамид натрия,и доведенных до метки водой.
Оптическую плотность полученных стандартных растворов измеряют на спектрофотометре при длине волны 261 нм в кювете с толщиной слоя 10 мм.
В качестве раствора сравнения используют воду.
По результатам измерений построить градуировочный график, рассчитать среднее значение удельного показателя поглощения ( ).
Для исследуемого раствора рассчитать концентрацию (Х) сульфацетамид натрия в процентах тремя способами, используя следующие формулы:
1.  ,
,
2.  ,
,
3. 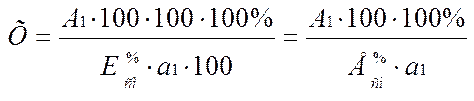 ,
,
где A1 и A0 – оптические плотности исследуемого раствора и ГСО;
а1 и а0 – точные массы исследуемого и ГСО сульфацетамид натрия (сульфацил-натрия), г;
100 – разведение, мл;
С0 – количество г ГСО в 1 мл раствора Б;
– удельный показатель поглощения раствора сульфацетамид натрия при длине волны 261 нм.
РАБОТА 2. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ПОДЛИННОСТИ, ЧИСТОТЫ КОЛИЧЕСТВЕННОГО СОДЕРЖАНИЯ ЦИАНОКОБАЛАМИНА В РАСТВОРЕ ВИТАМИНА В12 ДЛЯ ИНЪЕКЦИЙ (ФС)
Практические навыки
1. Освоить методики спектрофотометрического определения подлинности, чистоты, количественного содержания цианокобаламина в УФ- и видимой областях спектра.
2. Приготовить растворы анализируемого и стандартного образцов (РСО) цианокобаламина, согласно ФС, снять их электронные спектры в области длин волн от 210 до 800 нм и определить длины волн максимального поглощения (подлинность).
3. Определить поглощающие примеси, используя величины оптических плотностей при длинах волн 278, 361, 548 нм.
4. Определить количественное содержание цианокобаламина.
5. Написать отчёт.
РАБОТА 3. ФОТОЭЛЕКТРОКОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ТИМОЛА НА ОСНОВЕ РЕАКЦИИ НИТРОЗИРОВАНИЯ
Практические навыки
1. Освоить количественное определение фенолов (на примере тимола) на основе реакции Либермана методом фотоэлектроколориметрии.
2. Приготовить исследуемый раствор и серию стандартных растворов тимола, провести с ними реакцию образования 4-нитрозотимола.
3. Подобрать, используя один из стандартных растворов, подходящий светофильтр.
4. Определить величины оптических плотностей всех стандартных растворов при выбранном светофильтре.
5. Построить градуировочный график, определить область концентраций, подчиняющуюся закону Бугера-Ламберта-Бера, рассчитать .
6. Определить количественное содержание тимола в предложенном образце, используя:
а) градуировочный график;
б) значение ;
в) величину оптической плотности раствора Государственного стандартного образца (ГСО) тимола.
В качестве раствора ГСО тимола следует взять раствор из серии стандартных растворов, ближе расположенный к исследуемому по величине оптической плотности (см. Градуировочный график).
7. Написать отчет.
ВЫПОЛНЕНИЕ РАБОТЫ
Метод основан на способности тимола при взаимодействии с натрия нитритом в кислой среде образовывать 4-нитрозотимол, который в щелочной среде образует окрашенное в лимонно-желтый цвет соединение.
Около 0,0500 г (точная навеска) тимола помещают в мерную колбу вместимостью 100 мл, растворяют в 2 мл этанола и полученный раствор доводят водой до метки (раствор А). 0,5 мл полученного раствора помещают в мерную колбу вместимостью 100 мл, прибавляют 0,5 мл 10 % раствора кислоты уксусной, 0,5 мл 1 % раствора натрия нитрита. Через 30 мин прибавляют 4 мл 10 % раствора натрия гидроксида и доводят раствор до метки очищенной водой. Измеряют оптическую плотность полученного раствора на фотоэлектроколориметре при подобранном светофильтре (410 нм) в кювете с толщиной слоя 20 мм. В качестве контрольного раствора используют очищенную воду.
Параллельно проводят измерения серии стандартных растворов.
Построение градуировочного графика. Приготовить в мерных колбах вместимостью 100 мл серию растворов, состоящих из 0,1; 0,3; 0,5; 0,8; 1,0 мл 0,5 % раствора стандартного образца тимола и таких же реактивов (в том же количестве), которые добавлены к исследуемому раствору.
Оптическую плотность полученных стандартных растворов измерить в кювете с толщиной слоя 20 мм при том же светофильтре, что и в случае исследуемого раствора. По результатам измерений построить градуировочный график, рассчитать среднее значение удельного показателя поглощения ( ).
Для исследуемого раствора рассчитать концентрацию тимола (Х) в % тремя способами, используя следующие формулы:
1.  ;
;
2.  ;
;
3.  ,
,
где A1 и A0 – оптические плотности исследуемого раствора и ГСО тимола;
а1 и а2 – точные массы исследуемого и ГСО тимола, г;
0,5, 100, 100 – разведение, мл;
С0 – количество г ГСО тимола в1 мл раствора;
– удельный показатель поглощения тимола;
ℓ – толщина поглощающего слоя, см.
ГЛАВА 4. ГАЗОВАЯ ХРОМАТОГРАФИЯ
Хроматографией называется метод разделения смеси на составляющие ее компоненты, основанный на различной скорости движения веществ, непрерывно распределяющихся между двумя фазами – подвижной и неподвижной. Честь открытия метода принадлежит русскому ученому ботанику и физико-химику М.С. Цвету, который в 1903 году использовал для разделения растительных пигментов на их составляющие колонки, заполненные порошком мела.
Хроматография отличается от других двухфазных процессов разделения именно наличием неподвижной (стационарной) фазы с развитой поверхностью, что позволяет получить высокую эффективность на единицу длины слоя неподвижной фазы (НФ).
Подвижная фаза (ПФ) может быть газообразной или жидкой. Если ПФ газообразна, то процесс называется газовой хроматографии, а если жидкость, то такой процесс носит название жидкостной хроматографии.
Хроматографические методы дают возможность проводить качественный и количественный анализы лекарственных средств, изучать их физико-химические свойства, осуществлять контроль и автоматическое регулирование технологических процессов. Они активно применяются в научных исследованиях, в различных отраслях промышленности, в медицине, криминалистике, для контроля окружающей среды и т.д. Необычайно разнообразны анализируемые ими объекты: пищевые продукты, белки, лекарственные средства, микроорганизмы, нефть, газ, металлы, лунный грунт и атмосфера планет солнечной системы.
Наиболее широкое распространение из хроматографических методов аналитического и препаративного разделения многокомпонентных смесей получили газожидкостная (ГЖХ) и высокоэффективная жидкостная хроматография (ВЭЖХ), благодаря высокой чувствительности, эффективности, селективности, экспрессности, возможности автоматизации в сочетании с другими физико-химическими методами. Отличительной их особенностью является универсальность, т.е. возможность использования для разделения и определения твердых, жидких и газообразных неорганических и органических соединений в широком интервале концентраций.
Газовая хроматография является одним из широко применяемых аналитических методов. Она реализуется в двух модификациях в зависимости от агрегатного состояния неподвижной фазы:
Газовая адсорбционная;
2 – газожидкостная.
В первом случае неподвижной фазой является твердый сорбент, во втором – высококипящая жидкость, нанесенная в виде тонкой пленки на
твердый носитель. Анализируемое вещество в обоих случаях – газ.
В практике фармацевтического анализа широкое применение находит газожидкостная хроматография, которая была предложена в 1952 году английскими учеными А.Джеймсом и А.Мартином.
4.1. ГАЗОЖИДКОСТНАЯ ХРОМАТОГРАФИЯ
В основе газожидкостной распределительной хроматографии (ГЖХ) лежит различие в растворимости разделяемых веществ, на выбранном неподвижном растворителе в хроматографической колонке или более точно – различие коэффициентов их распределения между неподвижной жидкой фазой (НЖФ) и подвижной газовой фазой (ПГФ), газом-носителем.
Необходимыми условиями реализации этого метода являются летучесть компонентов смеси и их устойчивость при температуре разделительной колонки.
Анализируемые вещества (или смесь веществ) в газообразном состоянии смешиваются с потоком газа-носителя и проходят через колонку. В колонке находятся частички твердого носителя с тонким слоем высококипящей жидкости. Компоненты анализируемой смеси, растворяясь в этой жидкости распределяются между ПГФ и НЖФ в соответствии с коэффициентом распределения. После установления в первый момент равновесия между ПГФ и НЖФ газ вместе с нерастворившейся в НЖФ частью анализируемой пробы устремляется вглубь колонки, где также устанавливается равновесие. В то же время новая порция чистого газа-носителя вступает в равновесие с НЖФ, содержащей растворенные компоненты, и часть из них переходит в ПГФ.
Указанные процессы (последовательный переход из ПГФ в НЖФ и опять в ПГФ) совершаются до тех пор, пока молекулы анализируемых компонентов не пройдут через всю колонку. При этом менее растворимый в НЖФ компонент проходит через колонку быстрее, чем более растворимый, так как время его пребывания в стационарной фазе будет меньше. Принцип хроматографического разделения показан на рис.14.

Образец трехкомпонентной анализируемой смеси продувается с помощью газа-носителя через слой неподвижной жидкой фазы. Поскольку компоненты смеси обладают различной сорбируемостью, их движение в колонке будет замедляться по-разному. Чем больше сорбируемость молекул, тем сильнее будет их торможение и наоборот. Следовательно, компоненты смеси будут двигаться с разной скоростью. Через некоторое время вперед уйдет компонент С, как менее сорбирующийся, за ним компонент В и, наконец, компонент А, как более сорбирующийся и поэтому медленее движущийся. В момент (б) компоненты еще не полностью отделились друг от друга. Однако через некоторое время произойдет их полное разделение (в). На выходе из колонки состав выходящих порций газа фиксируется с помощью детектора и регистрируется на хроматограмме в виде пиков (рис.15).
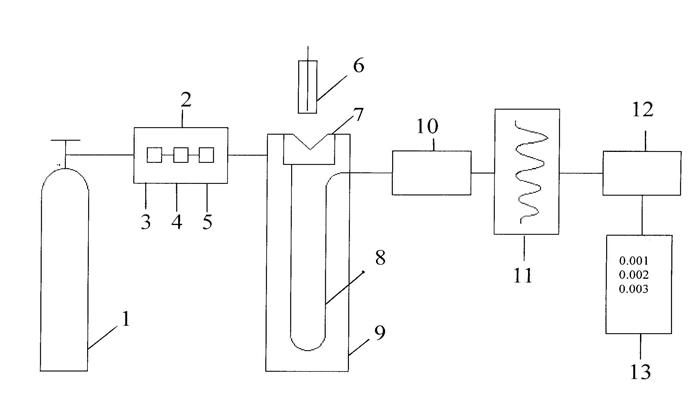 |
Блок-схема современного газового хроматографа представлена на рис.14.
Рис.15. Блок-схема газового хроматографа
1 – баллон с сжатым газом; 2 – блок подготовки газа-носителя; 3 – регулятор расхода газа; 4 – измеритель расхода газа; 5 – фильтр; 6 – микрошприц для введения пробы; 7 – испаритель; 8 – хроматографическая колонка; 9 – термостат; 10 – детектор; 11 – самописец; 12 – интегратор; 13 – цифропечатающее устройство.
Газ-носитель подается из газового баллона (1) через редуктор, а расход газа определяют с помощью регуляторов давления. Очистка газов от пыли, влаги, органических соединений осуществляется с помощью фильтров, установленных после баллона.
К газу-носителю предъявляется ряд требований: он должен быть инертным, достаточно чистым, иметь как можно меньшую вязкость, обеспечивать высокую чувствительность детектора, взрывобезопасным, доступным. Указанным требованиям удовлетворяют в основном гелий, азот, аргон.
Жидкие пробы вводят в поток газа-носителя обычно непосредственно путем впрыскивания из микрошприца (6) через мембрану, изготовленную из силиконовой самоуплотняющейся резины. Объем вводимой пробы зависит от типа детектора, количества НЖФ и диаметра колонки. Обычно объем смеси, анализируемой методом ГЖХ, составляет для жидкостей от сотых долей мкл до 10 мкл. Дозирование – одна из ответственных операций, и ошибки при ее выполнении составляют, как правило, большую часть погрешности анализа.
Испаритель(7) представляет собой нагреваемый до определенной температуры металлический блок с каналом для ввода и испарения жидкой пробы. В канал подается поток предварительно нагретого газа-носителя. Игла шприца с анализируемой жидкостью вводится через термостойкое уплотнение в канал испарителя. Введенная проба быстро испаряется и переносится потоком газа в хроматографическую колонку (8), где происходит сорбция. Обычно колонки изготавливают из стекла, кварца, нержавеющей стали, полимеров (чаще тефлона) в форме спирали. Различают 2 вида аналитических колонок: насадочные (набивные), капиллярные.
Насадочная колонка –разделительная колонка, внутренняя полость которой полностью заполняется инертным твердым носителем, покрытым тонкой пленкой нелетучего жидкого вещества (НЖФ). Обычно длина насадочных колонок колеблется от 1 до 5 м, а внутренний диаметр – от 2 до 4 мм.
Капиллярные колонкиполучили свое название от материала, из которого их изготавливают: капиллярных трубок с внутренним диаметром 0,1-0,5 мм и длиной от 10 до 100 м, выполненных чаще из плавленного кварца или стекла.
В качестве твердых носителей применяют материалы на основе кремнезема – диатомита и кизельгура (сферохромы, хромотоны, хезосорбы, цеолиты); фторуглеродных полимеров (тефлон, полихром), полистирола и сополимеров стирола и дивинилбензола (полисорбы) и др.
Неподвижные жидкие фазы. Для обеспечения селективности колонки важно правильно выбрать НЖФ. В качестве НЖФ применяют высококипящие жидкости: углеводороды и их смеси (вазелиновое масло, апиезоны), ДМФА, сложные эфиры и полиэфиры, силоксановые полимеры без функциональных групп и с некоторыми привитыми группами, полигликоли и др. Различают жидкие фазы трех типов: неполярные(насыщенные углеводороды и др.), умеренно полярные (сложные эфиры, нитрилы и др.) и полярные (полигликоли, гидроксиламины и др.)
В ГЖХ применяются силиконы различной полярности – от неполярных до сильнополярных. Каждая НЖФ характеризуется значением максимально допустимой рабочей температуры. Большая часть используемых в ГЖХ НЖФ не выдерживает высоких температур (свыше 200оС). Зная свойства НЖФ и природу разделяемых веществ, (класс, строение), можно достаточно быстро подобрать подходящую для разделения данной смеси селективную жидкую фазу.
Капиллярные колонкиразделяют по способу фиксации НЖФ на два типа: колонки с тонкой пленкой НЖФ (0,01–1 мкм) непосредственно на внутренней поверхности капилляров и тонкослойные колонки, на внутреннюю поверхность которых нанесен пористый слой (5–10 мкм) твердого вещества, выполняющего функцию носителя НЖФ.
В капиллярной колонке существенно уменьшается сопротивление потоку газа, поэтому появляется возможность увеличить длину колонки и повысить таким образом эффективность разделения.
Проба для анализов в капиллярной хроматографии уменьшается в 1000 раз и более, при этом существенно сокращается время анализа. направлять в колонку. Большая длина, малый диаметр капилляров обеспечивают высокую эффективность разделения смесей веществ, большую скорость хроматографического разделения, высокую чувствительность, селективность капиллярной хроматографии, являющейся разновидностью газожидкостной.
Выбор температуры разделения веществ имеет чрезвычайно важное значение для успеха анализа в целом. Температура колонок определяется главным образом летучестью пробы и может варьироваться до +3500С. Температуру колонки контролируют с точностью до нескольких десятых градуса и поддерживают с помощью термостата.
Температурный режим хроматографического процесса может быть различным.
При изотермическойхроматографии для каждой разделяемой смеси существует определенная оптимальная температура. Если компоненты сильно удерживаются на данной колонке, выходят из нее очень медленно или иногда не выходят совсем, то смесь веществ трудно разделить при постоянной температуре. Поэтому используют программирование температуры,чаще линейное, которое представляет собой повышение температуры колонки во время анализа с целью ускорения и обеспечения большей гибкости анализа
Выходящий из колонки газ-носитель вместе с разделенными компонентами поступает в измерительную ячейку детектора (10).
Следует подчеркнуть, что возможности хроматографа в основном определяются характеристиками используемого в нем детектора, который является наиболее ответственным узлом.
Детектор предназначен для обнаружения изменений в составе газа, прошедшего через колонку. Работа его основана на измерении таких физических и физико-химических свойств подвижной фазы и определяемых веществ, которые зависят от количества и природы веществ. Эти свойства детектор преобразует в электрический сигнал, который затем регистрируется самопишущим устройством (10). Для газовой хроматографии предложено около 20 типов детекторов, однако полный комплект современного универсального хроматографа включает не более 4-6 детекторов различных типов.
Наибольшее распространение в силу универсальности, превосходных характеристик и высоких эксплуатационных качеств получили детектор по теплопроводности (катарометр), действие которого основано на изменении теплопроводности газа-носителя в присутствии других веществ и пламенно-ионизационный детектор (ПИД),действие которогоосновано на изменении электропроводности пламени водородной горелки при прохождении через нее газовой смеси, выходящей из колонки.Они входят в состав почти всех хроматографов.
Электрический сигнал детектора непосредственно или через усилитель поступает на регистрирующий прибор. Для регистрации сигнала в большинстве случаев используют самопишущие потенциометры (милливольтметры). Перо регистратора записывает сигнал на движущейся диаграммной ленте или на экране монитора компьютера в виде хроматограммы.Каждое вещество на хроматограмме образует кривую, которую называют пиком(рис.16), при этом количество каждого компонента пропорционально его площади S.


При хорошо подобранных условиях разделения количество пиков на хроматограмме соответствует числу компонентов смеси. Обычно на оси абсцисс регистрируют время (с, мин), а на оси ординат – сигнал детектора (мВ или А).
Для соблюдения точности измерений необходимо всегда указывать температуру испарителя, колонки и детектора. В связи с этим у хроматографов имеются соответствующие блоки управления или компьютерные программы.Температура испарителядолжна быть достаточно высокой для того, чтобы обеспечить большую скорость испарения и достаточно низкой, чтобы исключить термическую деструкцию или изменение анализируемых соединений.
Температура колонкидолжна быть высокой для того, чтобы время анализа было небольшим и в то же время достаточно низкой, чтобы обеспечивалось требуемое разделение.
Температура детектора.Влияние температуры на характер работы детектора в значительной степени зависит от типа детектора. Однако общим правилом является необходимость поддержания температуры детектора и соединений между ним и колонкой достаточно высокой, чтобы исключить конденсацию анализируемых веществ жидкой фазы. Следует однако учесть, что чувствительность многих детекторов (например, катарометров) уменьшается с увеличением температуры, поэтому оптимальная температура лишь незначительно превышает температуру кипения наиболее высококипящего компонента.
Стабильность и связанная с ней максимальная чувствительность детектора по теплопроводности зависят от стабильности регулятора температуры детектора.
Ограничения в применении метода возникают из-за заметной летучести подавляющего большинства неподвижных жидких фаз при температуре разделительной колонки.
|
Просмотров 4418 |
|
|
