Главная Обратная связь
Дисциплины:
Архитектура (936)
Биология (6393)
География (744)
История (25)
Компьютеры (1497)
Кулинария (2184)
Культура (3938)
Литература (5778)
Математика (5918)
Медицина (9278)
Механика (2776)
Образование (13883)
Политика (26404)
Правоведение (321)
Психология (56518)
Религия (1833)
Социология (23400)
Спорт (2350)
Строительство (17942)
Технология (5741)
Транспорт (14634)
Физика (1043)
Философия (440)
Финансы (17336)
Химия (4931)
Экология (6055)
Экономика (9200)
Электроника (7621)

Производные диметилфенилацетамида
|
|
Кроме сложных эфиров п-аминобензойной кислоты местноанестезирующую активность проявляют производные диметилфенилацетамида (I). Среди них тримекаина гидрохлорид, лидокаина гидрохлорид, являющиеся производными диалкиламиноацетанилида (II) и бупивакаина гидрохлорид — производное пиперидинкарбоксамида (табл. 40.2).

Указанные лекарственные вещества имеют много общего в химической структуре. Это обусловливает общность их способов получения, свойств, испытаний и применения.
Синтез тримекаина осуществляют по схеме:

По такой же схеме синтезируют лидокаина гидрохлорид из 2,6-диметиланилина.
40.2. Свойства производных диметилфенилацетамида
| Лекарственное вещество | Химическая структура | Описание |
| Trimecaine Hydrochloride— тримекаина гидрохлорид |  2-(диэтиламино)-N-2’,4’,6’-триметилфенилацетамида гидрохлорида гемигидрат
2-(диэтиламино)-N-2’,4’,6’-триметилфенилацетамида гидрохлорида гемигидрат
| Белый или со слегка желтоватым оттенком кристаллический порошок. Т.пл. 139–142 °C |
| Lidocaine Hydrochloride— лидокаина гидрохлорид |  2-диэтиламино-2’,6’-ацетоксилидида
гидрохлорида моногидрат
2-диэтиламино-2’,6’-ацетоксилидида
гидрохлорида моногидрат
| Белый или почти белый кристаллический порошок, без запаха. Т.пл. 74–79 °C |
| Bupivacaine Hydrochloride— бупивакаина гидрохлорид | 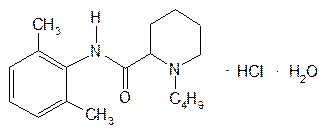 1-бутил-2’,6’-диметилфенил-2-пиперидинкарбоксамида гидрохлорида моногидрат
1-бутил-2’,6’-диметилфенил-2-пиперидинкарбоксамида гидрохлорида моногидрат
| Белый кристаллический порошок со специфическим запахом. Т.пл. 248 °C (с разложением) |
Они представляют собой белые или с желтоватым оттенком кристаллические вещества. Тримекаина и лидокаина гидрохлориды очень легко растворимы в воде, легко растворимы в этаноле и хлороформе, практически нерастворимы в эфире. Бупивакина гидрохлорид легко растворим в воде и этаноле, мало растворим в хлороформе и ацетоне.
Установить подлинность тримекаина гидрохлорида можно по ИК-спектру, снятому в вазелиновом масле. Он должен в области от 4000 до 700 см–1 иметь те же полосы поглощения, что и спектр стандартного образца. По ИК-спектру идентифицируют и бупивакаина гидрохлорид. УФ-спектр раствора тримекаина гидрохлорида в водно-спиртовой смеси, подкисленной хлороводородной кислотой имеет в области 250-300 нм максимумы поглощения при 262,5 нм и при 271 нм, а при 255 нм — минимум поглощения. Раствор бупивакаина гидрохлорида в 0,1 М растворе хлороводородной кислоты имеет максимум при 271 нм.
Тримекаина гидрохлорид даёт цветную реакцию с раствором ацетата меди (зеленое окрашивание). При добавлении к кристаллическому тримекаину гидрохлориду 2 капель концентрированной серной кислоты и 1 капли пергидроля появляется кроваво-красное окрашивание. Небольшая масса тримекаина гидрохлорида (0,001 г) при нагревании с несколькими каплями реактива Марки на водяной бане в течение 10 мин приобретает красное окрашивание. После добавления 10 капель воды появляется голубая флуоресценция.
Лидокаина гидрохлорид переводят в основание, растворяют в этаноле и испытывают на подлинность с помощью цветной реакции с раствором хлорида кобальта. Образуется синевато-зеленый осадок. Пикрат лидокаина гидрохлорида, промытый и высушенный после осаждения пикриновой кислотой, должен иметь температуру плавления около 230 °C.
При нагревании тримекаина и лидокаина гидрохлоридов с растворами щелочей или кислот образуются исходные продукты синтеза — 2,4,6-триметиланилин и 2,6-диметиланилин соответственно. Они вступают в реакции диазотирования и образования азокрасителя, характерные для первичных ароматических аминов:

Гидролитическое расщепление до диэтиламиноуксусной кислоты и 2,4,6-триметиланилина или 2,6-диметиланилина происходит и при пиролизе.
Для идентификации лидокаина гидрохлорида используют метод ГЖХ. Испытание выполняют при температуре детектора и испарителя 290°C (газ-носитель — азот) в аналитической колонке, заполненной Инертоном с 3% неподвижной жидкой фазой OV-17.
Испытание на подлинность и количественное определение бупивакаина гидрохлорида выполняют методом ВЭЖХ в одной пробе. Время удерживания около 6 минут. Неподвижная фаза — лихосорб, подвижная — смесь ацетонитрила с фосфатным буферным раствором (pH 6,8). Содержание рассчитывают с помощью внутреннего стандарта. Этим методом определяют и тримекаин, в т.ч. в биологических объектах.
Для отличия тримекаина гидрохлорида от других местноанестезирующих средств (тетракаина, прокаина) используют различные способы. Один из них основан на окислении тримекаина гидрохлорида при нагревании до 155–165 °C (масляная баня) в смеси сульфата меди (II) и концентрированной серной кислоты. После охлаждения смеси и добавления концентрированного раствора аммиака появляется синее окрашивание, а при УФ-облучении наблюдается красно-розовая флуоресценция.
Второй способ, рекомендованный ФС, заключается в выполнении микрокристаллоскопической реакции, которую проводят на предметном стекле, смешивая по 1 капле растворов тримекаина гидрохлорида, 0,1 М раствора дихромата калия и серной кислоты. Через 5–10 мин по краям смеси появляются кристаллы в виде игл, собранных в пучки или веточки.
Лекарственные вещества являются гидрохлоридами, поэтому дают положительную реакцию на хлорид-ион.
При испытании на чистоту в тримекаина гидрохлориде обнаруживают посторонние примеси методом ТСХ на пластинках Силуфол УФ-254, а в лидокаина гидрохлориде определяют наличие примеси первичных ароматических аминов (не более 0,01%) по цветной реакции с п-диметиламинобензальдегидом.
Примесь 2,6-диметиланилина в бупивакаина гидрохлориде определяют методом ГЖХ (с пламенно-ионизационным детектором), газ-носитель — азот, носитель 30% SE-30 на хромосорбе WAW.
Количественное определение тримекаина и лидокаина гидрохлоридов выполняют методом неводного титрования, используя в качестве растворителя смесь муравьиной кислоты и уксусного ангидрида (1:20). Титрантом служит 0,1 М раствор хлорной кислоты, индикатор кристаллический фиолетовый или судан III (лидокаин). Химизм этого процесса подробно рассмотрен на примере эфедрина гидрохлорида.
Бупивакаина гидрохлорид количественно определяют в среде ледяной уксусной кислоты в присутствии ацетата ртути (индикатор кристаллический фиолетовый).
Для определения тримекаина и лидокаина гидрохлоридов применимы также методы кислотно-основного титрования в водной среде (по связанной хлороводородной кислоте) и аргентометрии (по хлорид-иону).
Известны также способы спектрофотометрического определения тримекаина гидрохлорида в одном из максимумов поглощения и экстрационно-фотометрического анализа на основе реакции с салицилатным комплексом меди (II).
Лидокаина, тримекаина и бупивакаина гидрохлориды хранят по списку Б в сухом месте, в плотно укупоренной таре, предохраняющей от действия света, при комнатной температуре. Они разрушаются даже в отсутствие света, особенно во влажной атмосфере и при повышении температуры.
Тримекаина и лидокаина гидрохлориды применяют в качестве местноанестезирующих средств для инфильтрационной (0,25–0,5%-ные растворы) и проводниковой (1–2%-ные растворы) анестезии. Бупивакаина гидрохлорид — местноанестезирующее средство длительного действия — один из наиболее активных местных анестетиков. Выпускают его в виде 0,25 и 0,5%-ных растворов для инъекций в ампулах.
Исследования учёных показали, что местноанестезирующим действием обладают не только производные п-аминобензойной кислоты и диметилфенилацетамида, но и сложные эфиры карбоновых кислот гетероциклического ряда (бензофуранкарбоновой, тиофенкарбоновой и др.). К этой группе относится артикаина гидрохлорид (Articaine Hydrochloride). По химической структуре артикаина гидрохлорид (ультракаин) представляет собой гидрохлорид метилового эфира 4-метил-3[2-пропиониламинопропионамидо]-2-тиофенкарбоновой кислоты:

Молекула артикаина имеет сходство со структурой кокаина. Она включает элементы анестезиофорной части его молекулы, в частности, радикал 2-пропиониламинопропионамида. Это как бы раскрытая тропановая часть молекулы, а аналогичная кокаину сложноэфирная группа отличается лишь тем, что связана с тиофенкарбоновой кислотой.
Артикаина гидрохлорид хранят в защищённом от света месте при температуре до 25 °C.
Артикаина гидрохлорид — местноанестезирующее средство быстрого и относительно длительного действия при всех видах анестезии. Применяют его главным образом в стоматологии в виде 1 и 2%-ных растворов для инъекций по 1 мл. Ультракаин D-C — сочетание (в 1 мл) артикаина гидрохлорида (0,04 г) с адреналина гидрохлоридом (0,006 мг).
40.4. Производные амида пара-аминобензойной кислоты
В качестве лекарственных веществ применяются производные амида п-аминобензойной кислоты:

Много общего имеют в химической структуре прокаинамида гидрохлорид и метоклопрамида гидрохлорид с рассмотренными лекарственными веществами, производными п-аминобензойной кислоты (см).
Прокаинамида гидрохлорид синтезируют по схеме:

40.3. Свойства производных амида п-аминобензойной кислоты
| Лекарственное вещество | Химическая структура | Описание |
| Procainamide Hydrochloride— прокаинамида гидрохлорид (Новокаинамид) | 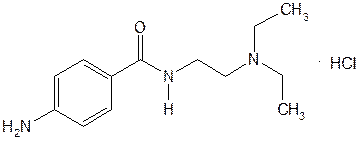 b-диэтиламиноэтиламида п-аминобензойной кислоты гидрохлорид
b-диэтиламиноэтиламида п-аминобензойной кислоты гидрохлорид
| Белый или белый со слегка кремоватым оттенком кристаллический порошок без запаха. Гигроскопичен. Т.пл. 165–169 °C |
| Metoclopramide Hydrochloride— метоклопрамида гидрохлорид (Церукал) |  4-амино-N-[2-(диэтиламино)этил]-2-метокси-5-хлорбензамида гидрохлорида моногидрат
4-амино-N-[2-(диэтиламино)этил]-2-метокси-5-хлорбензамида гидрохлорида моногидрат
| Белый или белый с желтоватым или кремоватым оттенком кристаллический порошок без запаха или почти без запаха |
Оба лекарственных вещества сходны по физическим свойствам (табл. 40.3). Они очень легко растворимы в воде, легко растворимы в этаноле, умеренно (метоклопрамид) или мало растворимы в хлороформе, практически нерастворимы в эфире.
Устанавливают подлинность прокаинамида и метоклопрамида гидрохлоридов по ИК-спектрам, которые сравнивают с соответствующим спектрами стандартных образцов. Рекомендуется также проводить измерение УФ-спектра раствора метоклопрамида в хлороводородной кислоте в области 230-350 нм. Максимумы поглощения должны находиться при 273 и 309 нм и иметь определённые величины оптических плотностей при заданной концентрации. Водные растворы прокаинамида гидрохлорида имеют один максимум поглощения при 278 нм, растворы в 0,02 М хлороводородной кислоте — при 275 нм, в 0,1 М растворе серной кислоты — при 224 нм. Согласно требованиям ФС 0,001%-ный спиртовой раствор метоклопрамида гидрохлорида в области 220-350 нм должен иметь максимумы поглощения при 212, 276, 311 нм, минимумы поглощения при 252 и 291 нм и плечо в интервале 225-234 нм.
Для испытания на подлинность прокаинамида и метоклопрамида гидрохлоридов могут быть использованы химические реакции, с помощью которых анализируют производные п-аминобензойной кислоты (см). Для подтверждения подлинности прокаинамида гидрохлорида используют реакцию образования азокрасителя. Он образует также дибром- или дииодпроизводные, изонитрилы, продукты конденсации с 2,4-динитрохлорбензолом и др.
Являясь гидрохлоридами, производные амида п-аминобензойной кислоты дают положительную реакцию на хлорид-ионы и выделяют осадки органических оснований под действием растворов гидроксида натрия. Они могут быть идентифицированы с помощью осадительных (общеалкалоидных) реактивов.
Прокаинамида гидрохлорид в растворе с концентрацией 0,01 г/мл в кислой среде образует с 2 каплями раствора перманганата калия (0,02 моль/л) фиолетовое окрашивание, которое быстро исчезает.
Подлинность прокаинамида гидрохлорида по МФ подтверждают цветной реакцией с гексацианоферратом (II) калия. В присутствии хлороводородной кислоты после нагревания образуется светло-зеленый осадок. Действуя на выделенное основание прокаинамида бензоилхлоридом, получают бензоилпрокаинамид:
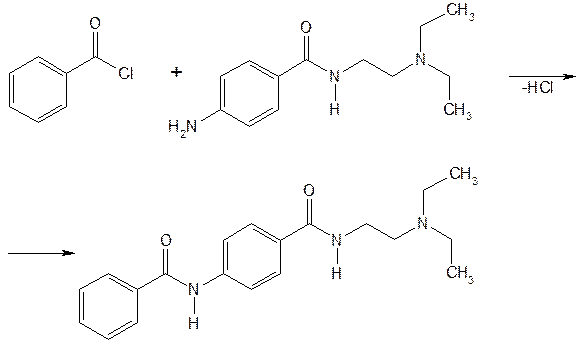
После перекристаллизации устанавливают температуру плавления. Она должна быть около 185°C.
Прокаинамида гидрохлорид, в отличие от прокаина, при нагревании с ванадатом аммония и концентрированной серной кислотой приобретает вишнево-красное окрашивание.
Метоклопрамида гидрохлорид с п-диметиламинобензальдегидом образует шиффово основание, имеющее жёлто-оранжевое окрашивание.
Наличие посторонних примесей в метоклопрамида гидрохлориде (по ФС — не более 1%) устанавливают методом ТСХ на пластинках Кизельгель 60F, используя смесь растворителей: бензол-этанол-раствор аммиака (45:15:1).
Количественное определение обоих лекарственных веществ может быть выполнено алкалиметрическим методом в водной среде по связанной хлороводородной кислоте или аргентометрическим методом по хлорид-иону. Для прокаинамида гидрохлорида ФС рекомендует также метод нитритометрии, который используют для количественного анализа производных п-аминобензойной кислоты.
Количественное определение прокаинамида гидрохлорида (по НД) выполняют методом неводного титрования в смеси уксусного ангидрида и ледяной уксусной кислоты (5:15). После нагревания до кипения прибавляют диоксан и ацетат ртути (II) и титруют 0,1 М раствором хлорной кислоты. Окончание титрования устанавливают потенциометрически.
Метоклопрамида гидрохлорид (по ФС) определяют методом неводного титрования по несколько изменённой методике. Навеску растворяют в ледяной уксусной кислоте, прибавляют уксусный ангидрид и точно отмеренные 4 мл 0,1 М раствора хлорной кислоты, оставляют в покое на 30 мин. Затем прибавляют раствор ацетата ртути (II), индикатор кристаллический фиолетовый и титруют 0,1 М раствором хлорной кислоты до изменения окраски индикатора. К расходу титранта прибавляют первоначально добавленные 4 мл. Параллельно выполняют в тех же условиях контрольный опыт, медленно титруя смесь растворов реактивов (без испытуемого вещества).
Разработан способ обнаружения и количественного определения прокаинамида гидрохлорида, основанный на использовании обращённо-фазового варианта ВЭЖХ с УФ-детектированием при длине волны 280 нм. Количественный анализ выполняют, используя подвижную фазу: метанол-буферный раствор с рН 4,0 (15:85). Внутренним стандартом служит прокаина гидрохлорид.
Газожидкостная хроматография использована в качестве подтверждающего метода для идентификации прокаинамида гидрохлорида, в т.ч. в биологических жидкостях. В лекарственных формах прокаинамида гидрохлорид количественно определяют методом УФ-спектрофотометрии, используя в качестве растворителя воду. Параллельно измеряют оптическую плотность стандартного и испытуемого растворов при длине волны 278 нм.
Хранят прокаинамида гидрохлорид (по списку Б) и метоклопрамида гидрохлорид в хорошо укупоренной таре, в сухом, защищённом от света месте, чтобы не допустить гидролиза. Прокаинамида гидрохлорид даже в отсутствие света постепенно разрушается во влажной атмосфере; при повышении температуры процесс гидролиза ускоряется.
Несмотря на сходство в химической структуре, метоклопрамида и прокаинамида гидрохлориды по фармакологическому действию различаются между собой. Метоклопрамида гидрохлорид успокаивает рвоту и икоту, вызванную различными причинами (наркоз, беременность и др.). Выпускают его в таблетках по 0,01 г и 0,5%-ные растворы в ампулах по 2 мл для инъекций. Прокаинамида гидрохлорид относится к антиаритмическим средствам. Хорошо всасывается из ЖКТ, частично ацетилируется в печени, образуя активный метаболит N-ацетилпрокаинамид. Назначают при расстройствах сердечного ритма в виде таблеток по 0,5-1,0 г или в вену по 5-10 мл 10%-ного раствора.
40.5. Производные n-аминосалициловой кислоты
n-Аминосалициловая кислота (ПАСК) впервые описана в 1902 г., однако ее фармакологическое действие было установлено значительно позже (только в 40-х годах). n-Аминосалициловая кислота и ее производные обладают бактериостатической активностью в отношении микобактерий туберкулеза. Применяют в медицине натрия пара-аминосалицилат (табл. 40.4).
Промышленный метод синтеза основан на превращении нитробензола в м-аминофенол реакцией электрофильного замещения (сульфирования), гидрирования нитрогруппы в аминную и замещения сульфогруппы на гидроксильную. Затем реакцией Кольбе-Шмидта (подобно получению салициловой кислоты) карбоксилируют м-аминофенол:

Полученную натриевую соль ПАСК Na очищают переосаждением и выделяют после кристаллизации в виде дигидрата.
Натриевая соль n-аминосалициловой кислоты представляет собой кристаллическое вещество (табл. 40.4), легко растворима в воде, трудно растворима в этаноле.
40.4. Свойства натрия пара-аминосалицилата
| Лекарственное вещество | Химическая структура | Описание |
| Natrii paraaminosalicylas— натрия парааминсалицилат (ПАСК Na) |  натриевая соль n-аминосалициловой
кислоты
натриевая соль n-аминосалициловой
кислоты
| Белый или белый со слегка желтоватым или розоватым оттенком мелкокристаллический порошок. Т. пл. 122 °C |
При установлении подлинности обнаруживают наличие иона натрия у натрия пара-аминосалицилата. УФ-спектры водных растворов имеют два максимума поглощения. Соотношение оптических плотностей 0,001%-ного водного раствора при длинах волн 265 и 299 нм должно быть в пределах 1,50-1,56, а величины удельных показателей поглощения равны соответственно 736 и 483.
Присутствие в молекуле п-аминосалицилат-иона фенольного гидроксила позволяет применять для его идентификации реакции на фенолы (см). При действии бромной воды или бромат-бромидной смеси из растворов выделяются осадки белого или желтоватого цвета. С хлороформом и щелочью образуются ауриновые красители желтого цвета. При взаимодействии с ксантгидролом натрия пара-аминосалицилат приобретает темно-зеленое окрашивание.
Наличие в молекуле фенольного гидроксила обусловливает положительную реакцию с раствором хлорида железа (III). Образуются соединения, окрашенные в кислой среде в фиолетовый цвет. После выполнения этой реакции из окрашенного раствора не должен выпадать осадок в течение трех часов. Образование осадка свидетельствует о примеси в натрия пара-аминосалицилате фармакологически неактивного м-аминосалицилата натрия:

Отличить натрия пара-аминосалицилат от м-аминосалицилата можно также реакцией с концентрированной серной кислотой и несколькими крупинками нитрита натрия. При нагревании появляется фиолетовое окрашивание, которое сохраняется после добавления избытка водного раствора аммиака. Окрашенное соединение извлекается амиловым спиртом, слой которого приобретает карминово-красный цвет и красную флуоресценцию.
Натрия пара-аминосалицилат, имея в молекуле незамещенную первичную ароматическую аминогруппу, образует с b-нафтолом азокраситель красного цвета:

За счет наличия в молекуле первичной ароматической аминогруппы натрия пара-аминосалицилат дает положительные реакции образования шиффовых оснований, конденсации с 2,4-динитрохлорбензолом (желтое окрашивание).
Кроме рассмотренных испытаний, натрия пара-аминосалицилат дает характерные цветные реакции с солями тяжелых металлов катионы которых взаимодействуют с карбоксильной и гидроксильной группами. Например, с ионом меди (II) он приобретает травянисто-зеленое окрашивание.
При испытании на чистоту натрия пара-аминосалицилата устанавливают наличие примеси м-аминофенола (промежуточный продукт синтеза). Испытание основано на извлечении м-аминофенола диэтиловым эфиром и установлении допустимых его количеств с помощью реакции образования азокрасителя с n-нитроанилином:
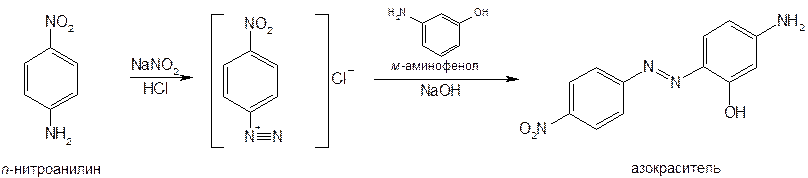
Количественное определение натрия пара-аминосалицилата можно выполнить различными методами. ФС рекомендует нитритометрию с внешним индикатором (иодкрахмальной бумагой). Определение броматометрическим и иодхлорометрическим методами подобно определению производных n-аминобензойной кислоты (см.). Известен способ спектрофотометрического определения натрия пара-аминосалицилата при длине волны 265 нм (растворитель вода).
Хранят натрия пара-аминосалицилат в хорошо укупоренной таре, предохраняющей от действия света, в сухом, защищенном от света месте, чтобы не допустить образования примесей продуктов разложения.
Натрия пара-аминосалицилат применяют внутрь для лечения различных форм туберкулеза по 2,0–3,0 г.
40.6. Производные м-аминобензойной кислоты

Применяют в медицине триомбрин (кислоту амидотризоевую) и лекарственную форму триомбраст 60% или 76% для инъекций (табл. 40.5).
Синтез кислоты амидотризоевой осуществляют из 3,5-диаминобензойной кислоты:

40.5. Свойства производных м-аминобензойной кислоты
| Лекарственное вещество (препарат) | Химическая структура (состав) | Описание |
| Amidotrizoic acid— кислота амидотризоевая (Триомбрин) |  3,5-диацетиламино-2,4,6-трииодбензойная кислота
3,5-диацетиламино-2,4,6-трииодбензойная кислота
| Белый кристаллический порошок без запаха. Т. пл. 260 °C (с разложением) |
| Triombrastum 60% et 76% pro injectionibus— триомбраст 60% или 76% для инъекций | Раствор смеси натриевой и метилглюкаминовой солей 3,5-диацетиламино-2,4,6-трииодбензойной кислоты | Прозрачная, бесцветная или светло-жёлтого цвета жидкость с pH 6,5-7,7 |
Кислота амидотризоевая легко растворима в растворах едких щелочей, мало — в этаноле, очень мало в воде, практически нерастворима в эфире и хлороформе. Триомбраст готовят, растворяя указанные в ФС определённые количества кислоты амидотризоевой, N-метилглюкамина, гидроксида натрия и динатриевой соли этилендиаминтетрауксусной кислоты в воде для инъекций.
Подлинность кислоты амидотризоевой подтверждают по ИК-спектру (он должен соответствовать спектру сравнения) и по УФ-спектру в 0,1 М растворе гидроксида натрия. В области 220-280 нм она имеет максимум поглощения при длине волны 238 нм (триомбраст в тех же условиях — при 237 нм).
По МФ подлинность кислоты амидотризоевой устанавливают реакцией на первичные ароматические амины (образование азокрасителя). Поскольку обе первичные ароматические аминогруппы ацетилированы, вначале осуществляют гидролиз, затем получают бис-диазосоединение. После взаимодействия с b-нафтолом выпадает красно-фиолетовый осадок.
При прокаливании в тигле кислоты амидотризоевой с серной кислотой разведённой, или при нагревании триомбраста с той же кислотой, но концентрированной, выделяются фиолетовые пары иода.
Для обнаружения иода кислоту амидотризоевую кипятят с обратным холодильником в растворе гидроксида натрия в присутствии цинковой пыли:

Образовавшийся иодид-ион обнаруживают реакцией с нитратом серебра; выпадает жёлтый творожистый осадок:
I– + AgNO3 ¾® AgI¯ + NO3–
К фильтрату (после отделения осадка) прибавляют 0,1 М раствор нитрита натрия. Выпадает коричневый с красноватым оттенком осадок (отличие от билигноста, иодамида).
Наличие кислоты амидотризоевой в триомбрасте устанавливают методом ТСХ на пластинке Силуфол УФ-254 в системе метанол-хлороформ-25% раствор аммиака (10:20:2). Сравнивают со свидетелем (кислотой амидотризоевой). Пятно от испытуемой пробы должно находиться на том же уровне, что и пятно от стандарта.
В кислоте амидотризоевой и триомбрасте определяют содержание примесей иода и неорганических иодидов. Испытание основано на извлечении этих веществ толуолом, слой которого не должен быть окрашенным (иод). После добавления 2%-ного раствора нитрита натрия слой толуола не должен окраситься в розовый цвет. В триомбрасте допускается не более 0,02% иодидов (в пересчёте на кислоту амидотризоевую). Определяют также наличие примеси соединений с открытой аминогруппой. В основе испытания лежит использование реакции диазотирования и образования азокрасителя (с нитритом натрия, сульфаминовой кислотой и a-нафтолом). В кислоте амидотризоевой допускается содержание соединений с открытой аминогруппой не более 0,015%, а в триомбрасте не более 0,05% (в пересчёте на кислоту амидотризоевую).
Для определения содержания органически связанного иода в кислоте амидотризоевой ранее использовался только деструктивный метод элементного анализа. Проведёнными исследованиями (Боковикова Т.Н.) была показана необходимость дополнительного использования недеструктивного метода определения по карбоксильной группе. Поэтому в ФС включены две методики. Одна из них основана на определении содержания иода в кислоте амидотризоевой после разрушения органической части молекулы (раствором гидроксида натрия в присутствии цинковой пыли) до образования эквивалентного количества иодид-ионов (химизм см. выше). Иодиды отделяют и титруют в фильтрате раствором нитрата серебра, устанавливая эквивалентную точку визуально (с индикатором эозинатом натрия) или потенциометрически с серебряным индикаторным электродом. Эту же методику ФС рекомендует для определения содержания кислоты амидотризоевой или иода в триомбрасте.
Вторая методика количественного определения кислоты амидотризоевой основана на использовании кислотных свойств её раствора в метаноле. В качестве титранта применяют 0,1 М раствор гидроксида натрия и смешанный индикатор. Параллельно проводят контрольный опыт. Расхождение между результатами анализа двумя методиками не должно превышать 1%. Использование двух указанных методик позволяет получить достаточно точную и объективную информацию о количественном содержании кислоты амидотризоевой по фармакологически активной части молекулы.
Описана также методика количественного определения кислоты амидотризоевой, основанная на использовании спектрофотометрии в УФ-области при длине волны 237 нм. В качестве растворителя используют 0,1 М раствор гидроксида натрия. Расчёты выполняют по значению оптической плотности РСО, устанавливая содержание кислоты амидотризоевой и иода в 60% или 76%-ном триомбрасте. В триомбрасте определяют также поляриметрическим методом содержание N-метилглюкамина, используя для расчётов значение удельного вращения его растворов.
Кислоту амидотризоевую хранят в хорошо укупоренной таре, защищающей от действия света. Ампулы с триомбрастом (по 20 мл) хранят по списку Б в защищённом от света месте, чтобы не допустить разложения с выделением иода.
Применяют в качестве рентгеноконтрастного средства триомбраст — водные 60% и 76%-ные растворы смеси натриевой и метилглюкаминовой солей кислоты амидотризоевой. Вводят внутривенно или в полость от 20 до 80 мл при рентгенологическом исследовании кровеносных сосудов, сердца, почек, мочевыводящих путей.
ГЛАВА 41.
|
Просмотров 10990 |
|
|
